L3 Pharmacokinetics
Properties of an ideal drug
- Safety: Fewer side effects or lower toxicity
- Effectiveness: Better therapeutic effects
- Selectivity: Target to desired sites or molecules
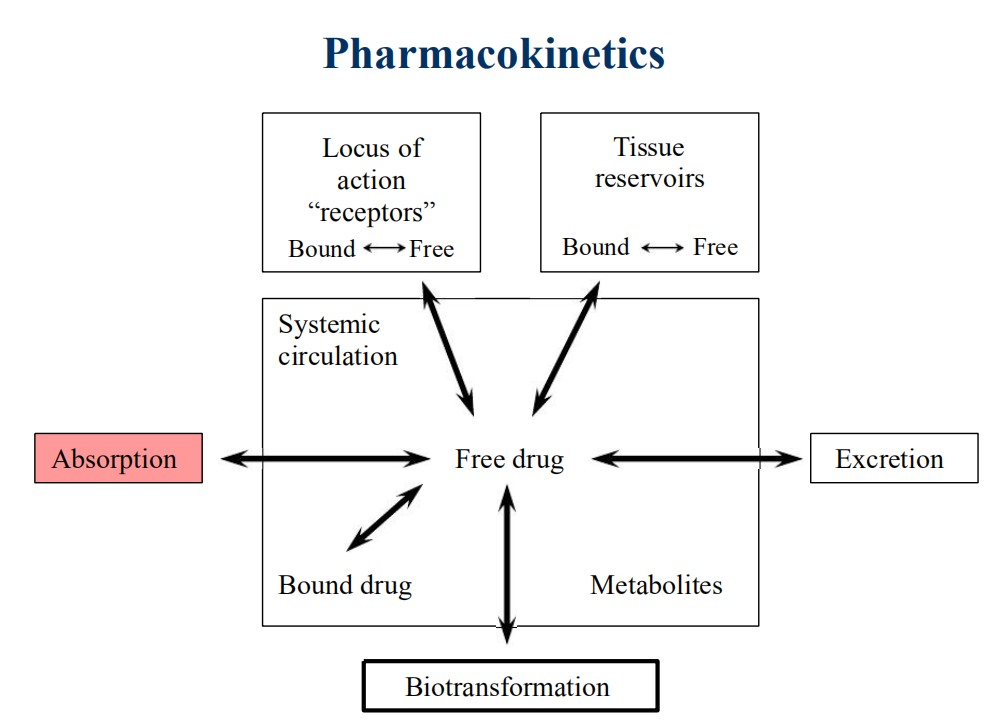
Pharmacokinetics & Pharmacodynamics
- PD governs the concentration-effect part of the interaction, whereas pharmacokinetics deals with the dose-concentration part
- The PK processes of ADE determine how rapidly and for how long the drug will appear at the target organ
Pharmacokinetic Process-ADME
- Absorption
- Distribution
- Metabolism
- Excretion
- It is also called ADME process
- Elimination is the sum of M and E
- Elimination Of Drugs
- Metabolism: Liver
- Excretion: Kidney, Liver (bile), Lungs
一、Drug administration
Route and form in which drug is delivered to body
Oral administration
Oral administration-most common route
Governed by:
- surface area for absorption, blood flow, physical state of drug, concentration.
- occurs via passive process.
- In theory: weak acids optimally absorbed in stomach, weak bases in intestine
- In reality: the overall rate of absorption of drugs is always greater in the intestine
1. Subject to First Pass Effect
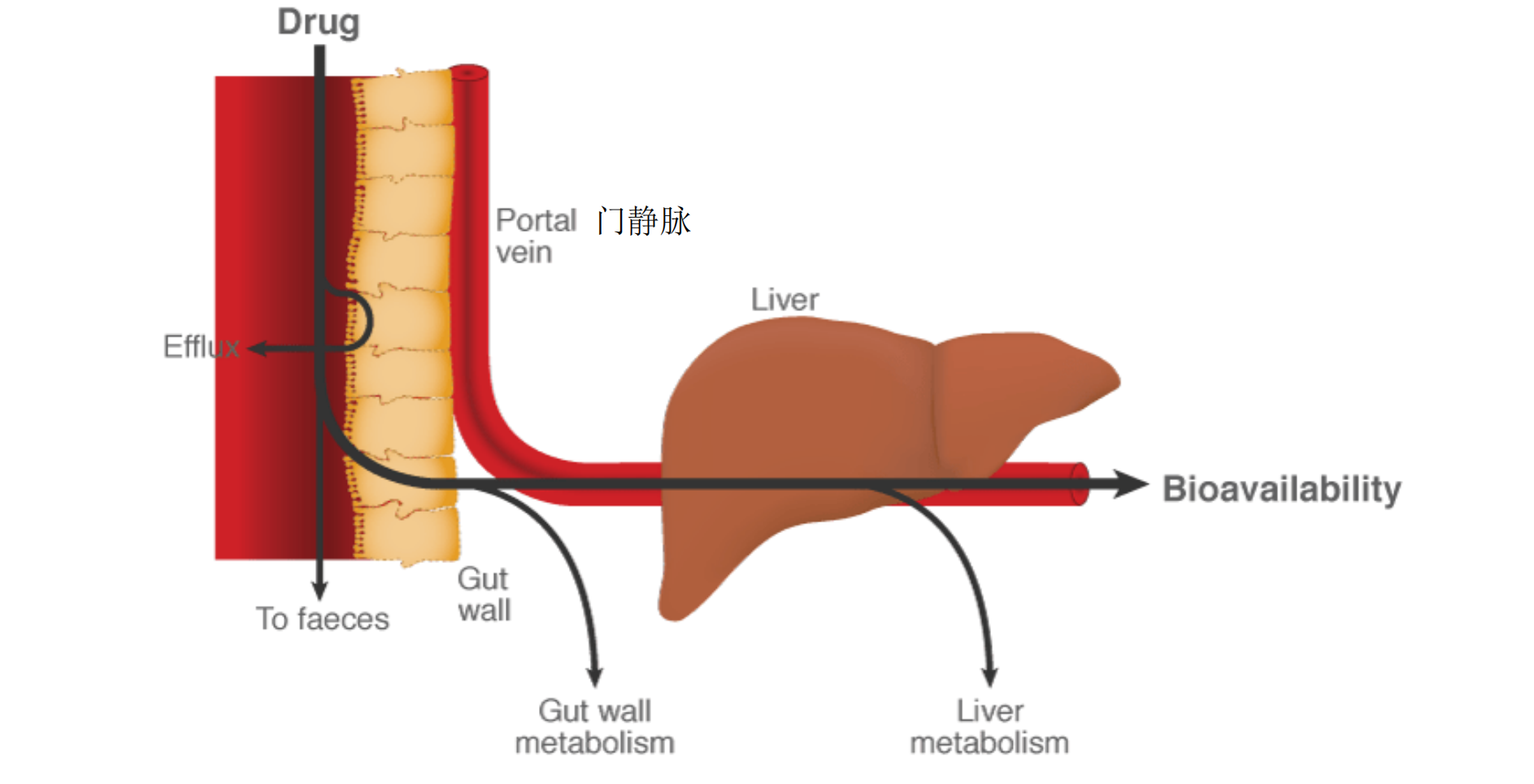
- pass through liver before reaching circulation
- undergo metabolism by liver
2. Rate of Appearance in Blood
Dependent on rate of dissolution
Rate of absorption from GI tract
- timed release
- dissolve at different rates
3. Forms of Oral Drugs
- liquids: syrups, elixirs
- suspensions
- powders
- pills: capsules, tablets

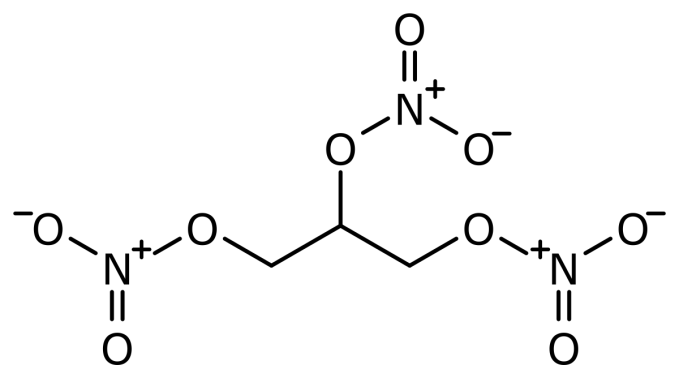
Buccal/Sublingual 颊部/舌下给药
sublingual (SL) = under tongue
absorbed through oral mucus membranes in mouth
- buccal = cheek
1. Sublingual Administration 舌下给药
Absorption from the oral mucosa has special significance for certain drugs despite the small surface area.
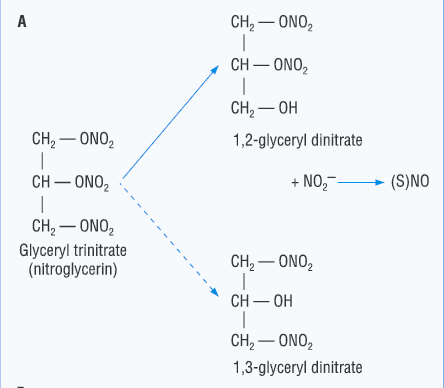
- Nitroglycerin: nonionic, very lipid Soluble, releases NO for vasodilation
- Because of venous drainage(静脉引流) into the superior vena cava(上腔静脉), this route “protects” it from first-pass liver metabolism.
- nonionic and lipid soluble 保证了能够快速穿过细胞膜
- 舌下给药的方式避免了通过肝脏,避免了肝脏对药物的消除作用,从而使得药物的利用效率更高
- 适用于需要快速比较紧急或避免肝脏的(首关)消除的方法
Parenteral 注射

Route other than alimentary canal
- intradermal (ID) 皮内注射
- subcutaneous (SC or SQ) 皮下注射
- intramuscular (IM) 肌内注射
- intravenous (IV) 静脉注射
1. Intravenous (IV)
directly into vein
- rapid onset of drug actions
- Fastest
2. Intramuscular
Rapid rate of absorption from aqueous solution, depending on the muscle.
Perfusion of particular muscle influences the rate of absorption
Slow & constant absorption of drug when injected in an oil solution or suspension
3. Subcutaneous
Slow and constant absorption
Slow-release pellet may be implanted
Drug must not be irritating
Topical: on the skin
not absorbed in appreciable amounts
- have local effects: (keep in mind large surface area)
absorbed
- transdermal: usually slowly over a day
Example not absorbed:

- A cream to treat Eczema
Example: absorbed

Example: Nitroglycerin
Inhalation
systemic drugs: intended to be absorbed into blood

local drugs: designed to act on lung tissue
Intraarterial Administration 动脉注射
Occasionally a drug is injected directly into an artery to localize its effect to a particular organ, e.g., for liver tumors, head/neck cancers.
Requires great care and should be reserved for experts.
Intrathecal
Necessary if the blood-brain barrier and blood-CSF barrier impede entrance into the CNS.

Used for local or rapid effects of drugs on the meninges or cerebrospinal axis, as in spinal anesthesia or acute CNS infections.
Rectal/Vaginal
**Suppositories (**栓剂)
- Vaginal: usually not absorbed
- Local Effect

- Rectal: absorbed
- May be useful when oral administration is precluded by vomiting or when the patient is unconscious.
- some first pass effect, approximately 50% of the drug that is absorbed from the rectum will bypass the liver.

Choice of route of drug administration
Consider factors
- Concentration in blood
- Rapidity of onset
- Duration of effects
- Magnitude of effects
- Amount delivered
二、Drug Absorption
Process of drug leaving site of administration
1. Important Properties Affecting Drug Absorption
Chemical properties
- acid or base
- degree of ionization
- polarity
- molecular weight
- molecular is small → Lipid soluble
- lipid solubility
- partition coefficient
Physiologic variables
- gastric motility 胃蠕动
- pH at the absorption site
- area of absorbing surface
- blood flow
- Pre-systemic elimination
- ingestion w/wo food
2. Influenced by
size & thickness of absorbing surface
- GI tract
- large with single cell layer
- small bowel > large bowel
- large with single cell layer
Surface
- Lung
- Large with single cell layer
- Mucus membranes
- Surface small with multiple cell layers
- Skin
- Surface large with multiple cell layers
blood supply to surface
- GI, lung, mucus membranes
- good blood supply
- skin: variable
3. Factors Influencing Absorption
- interaction of drug with surface characteristics
- e.g. skin: low in water and lipid
- amount of time in contact with surface
- concentration gradient
Cell Membranes
The character of biological membrane
- Be composed of phospholipids and proteins
- lipid soluble molecule can cross it easily
- Has aqueous channel
- water and liquid soluble molecule with little molecule weight can cross it easily
1. Membranes and Absorption
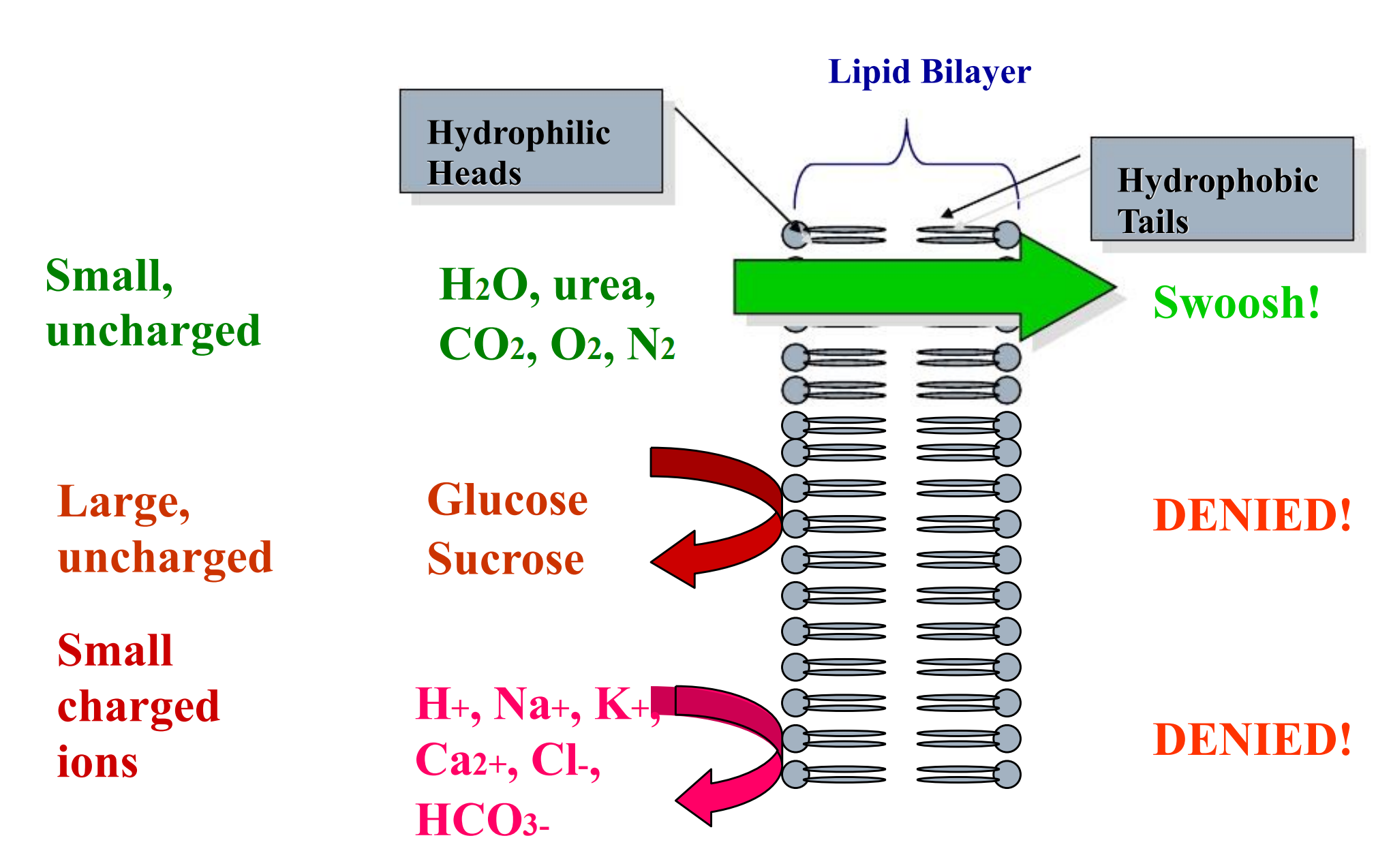
Aqueous diffusion
Water-soluble, small molecular mass drugs
The force of it is the liquid static pressure or osmotic pressure across the membrane
Through aqueous channels
(1) Molecular Size
In general, smaller molecules diffuse more readily across membranes than larger ones (because the diffusion coefficient is inversely related to the sq. root of the MW). This applies to passive diffusion but NOT to specialized transport mechanisms (active transport, pinocytosis)
- tight junction: MW <200 for diffusion
- large fenestrations in capillaries: MW 20K-30K.
Lipid diffusion
The lipid-soluble drug molecule penetrates along a concentration gradient by virtue of its solubility in the lipid membrane
(1) Oil: Water Partition Coefficient
The greater the partition coefficient, the higher the lipid-solubility of the drug, and the greater its diffusion across membranes.
A non-ionizable compound (or the non-ionized form of an acid or a base) will reach an equilibrium across the membrane that is proportional to its concentration gradient.
Ionization
Most drugs are small (MW < 1000) weak electrolytes (acids/bases). This influences passive diffusion since cell membranes are hydrophobic lipid bilayers that are much more permeable to the non-ionized forms of drugs.
The fraction of drug that is non-ionized depends on its pKa, and the local biophase pH…
ionized = polar = water-soluble
non-ionized = less polar = more lipid-soluble
- Think of an acid as having a carboxyl: COOH / COO-
- Think of a base as having an amino: NH3+ / NH2
For both acids and bases, pKa = acid dissociation constant, the pH at which 50% of the molecules are ionized.
Example:
- weak acid = aspirin (pKa 3.5)
- weak base = morphine (pKa 8.0)
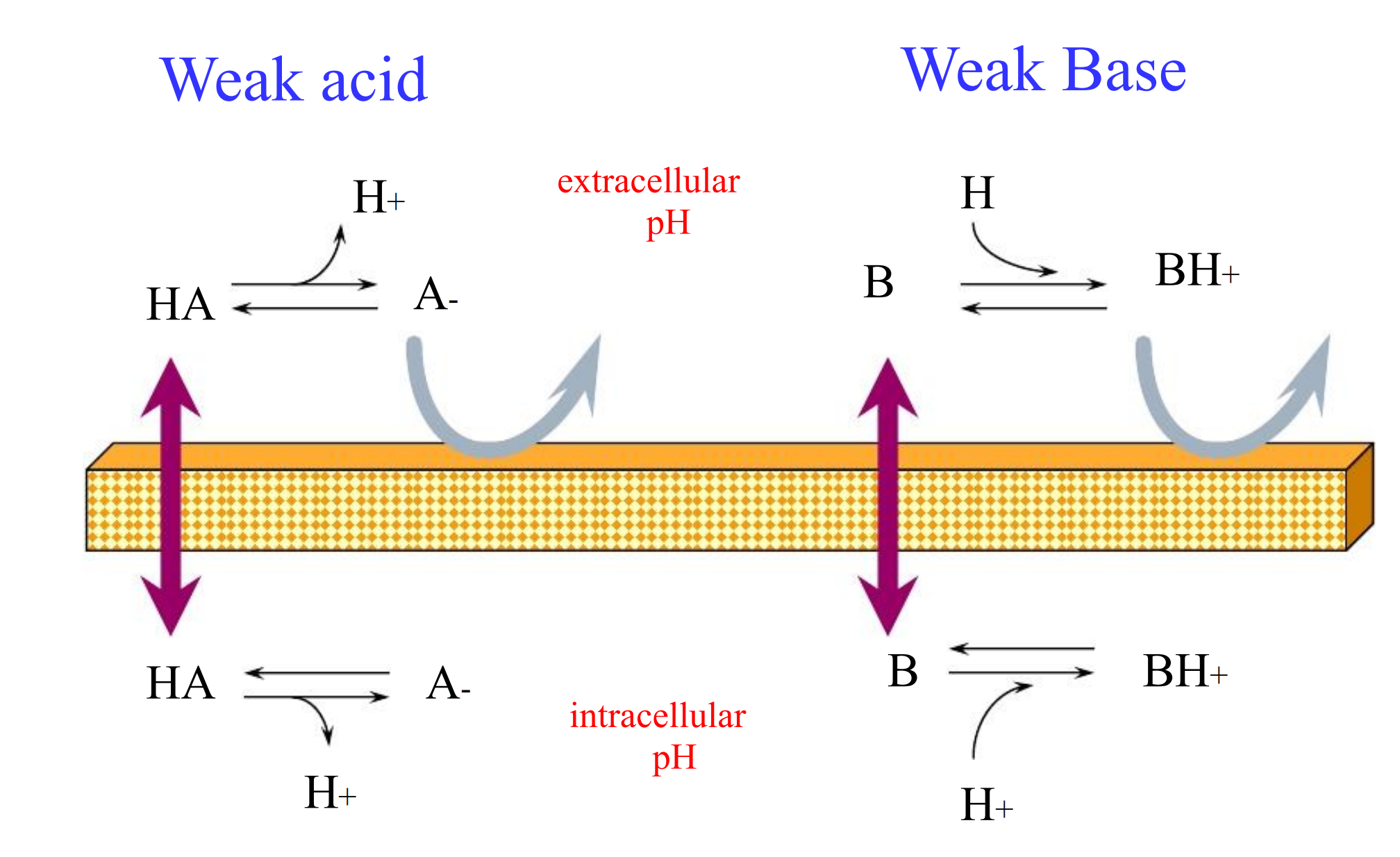
- The pH on each side of the membrane determines the equilibrium on each side
(1) Henderson-Hasselbalch Eqn
$$
\log{\frac{\text{protonated}}{\text{unprotonated}}} = pK_{a} - pH
$$


Moral of the story
- Acids are increasingly ionized with increasing pH (basic environment)
- Bases are increasingly ionized with decreasing pH (acidic environment)
Acidic drugs are best absorbed from acidic environments
- Basic drugs are best absorbed from basic environments
Certain compounds may exist as strong electrolytes. This means they are ionized at all body pH values. They are poorly lipid soluble.
**Example: **
- strong acid = glucuronic acid derivatives of drugs.
- strong base = quarternary ammonium compounds such as acetylcholine
2. Membrane Transfer
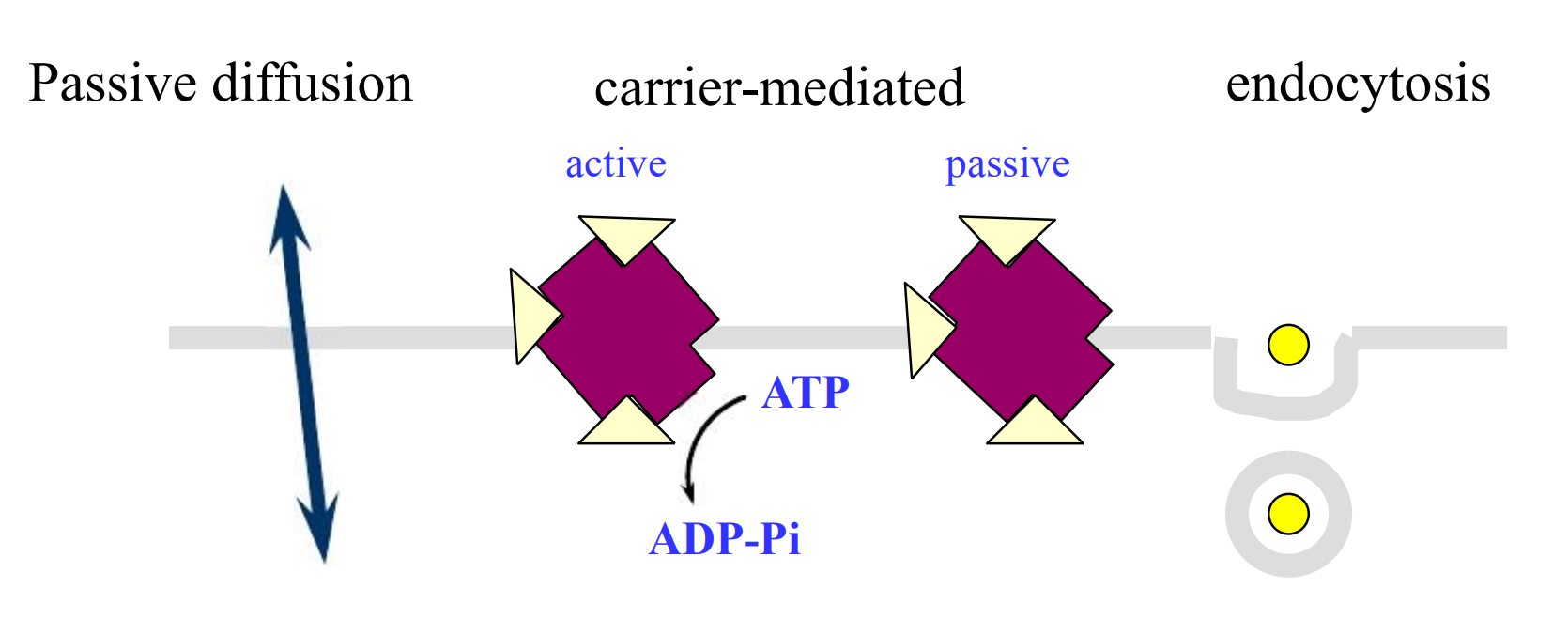
Special carriers
Substances that are important for cell function and too large or too insoluble in lipid to diffuse passively through membranes, eg, peptides, amino acids, glucose.
These kind of transport, unlike passive diffusion, is saturable and inhibitable
- Active transport is characterized by a requirement for energy
- Facilitated diffusion needs no energy
Active Transport
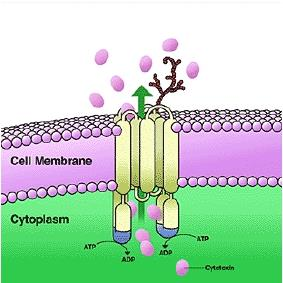
Occurrence:
- neuronal membranes, renal tubule cells, hepatocytes
Characteristics:
- carrier-mediated
- selectivity
- competitive inhibition by congeners
- energy requirement
- saturable
- movement against concentration gradient
Facilitated Diffusion
This is a carrier-mediated process that does NOT require energy. In this process, movement of the substance can NOT be against its concentration gradient.
Necessary for the transport of endogenous compounds whose rate of movement across membranes by simple diffusion would be too slow
Endocytosis, Exocytosis, Internalization
Endocytosis (or pinocytosis): a portion of the plasma membrane invaginates and then pinches off from the surface to form an intracellular vesicle.
Example: This is the mechanism by which thyroid follicular cells, in response to TSH, take up thyroglobulin (MW > 500,000).
三、Drug Distribution
via circulation to sites of action sites of elimination
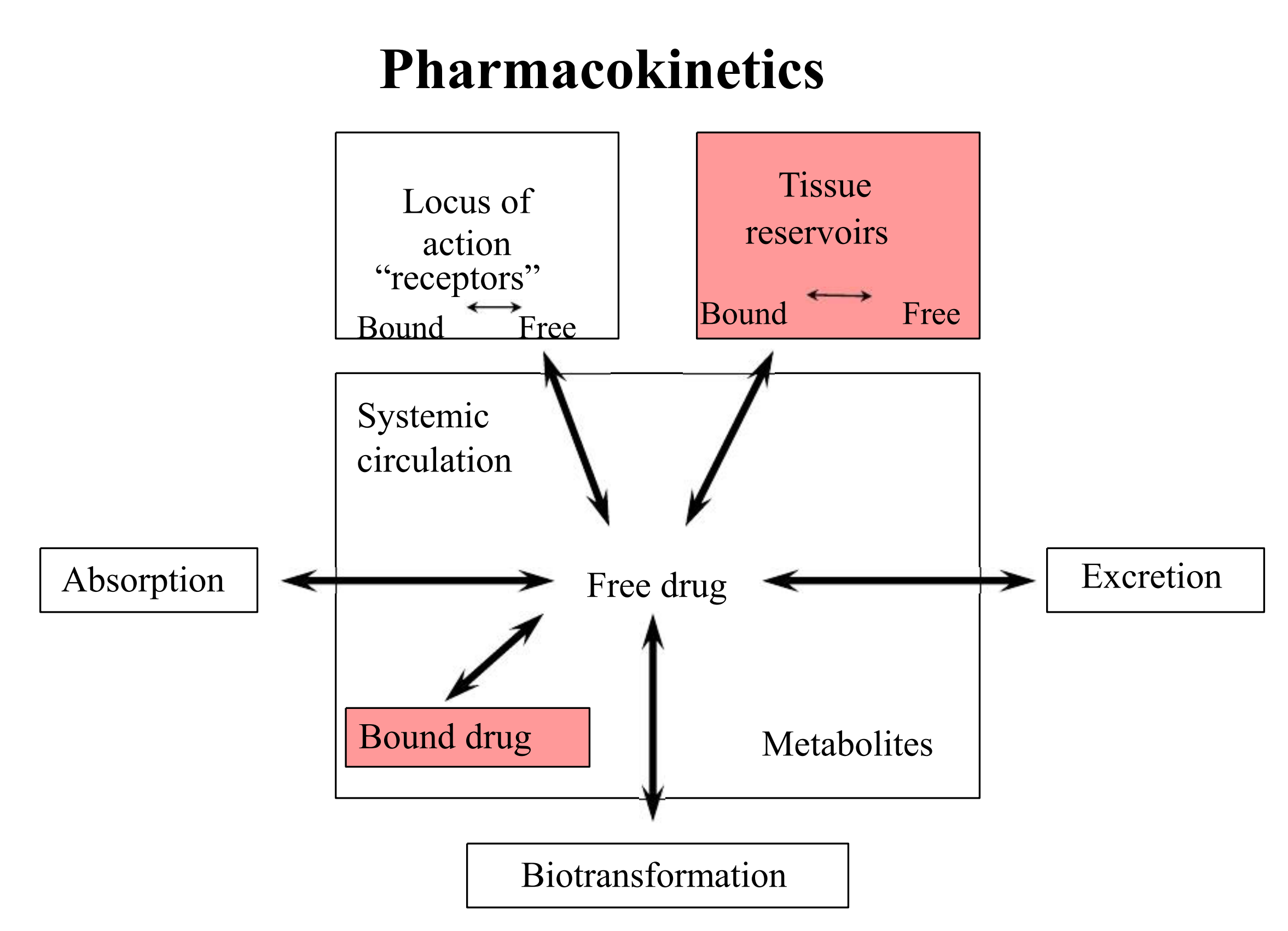
Phases of Distribution
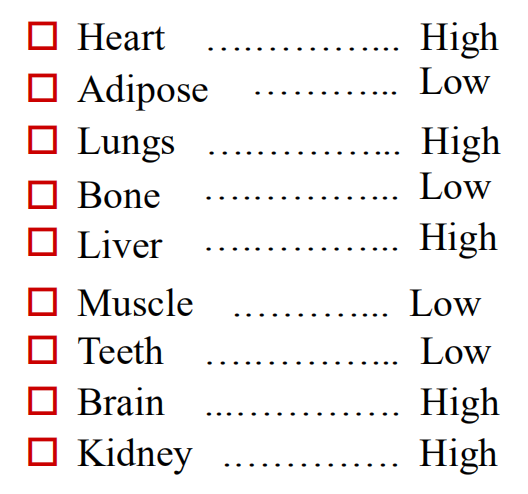
first phase
- reflects cardiac output and regional blood flow. Thus, heart, liver, kidney & brain receive most of the drug during the first few minutes after absorption.
next phase
- delivery to muscle, most viscera(内脏), skin and adipose is slower, and involves a far larger fraction of the body mass.
Drug Reservoirs
Body compartments where a drug can accumulate are reservoirs. They have dynamic effects on drug availability.
plasma proteins as reservoirs (bind drug)
cellular reservoirs
- Adipose (lipophilic drugs)
- Bone (crystal lattice)
- Transcellular (ion trapping)
1. Adipose Reservoir
Many lipid-soluble drugs are stored in fat. In obesity, fat content may be as high as 50%, and in starvation it may still be only as low as 10% of body weight.
2. Bone Reservoir
Tetracycline antibiotics (and other divalent metal ion-chelating agents) and heavy metals may accumulate in bone. They are adsorbed onto the bone-crystal surface and eventually become incorporated into the crystal lattice
Bone then can become a reservoir for slow release of toxic agents (e.g., lead, radium) into the blood
3. GI Tract as Reservoir
Weak bases are passively concentrated in the stomach from the blood because of the large pH differential.
Some drugs are excreted in the bile in active form or as a conjugate that can be hydrolyzed in the intestine and reabsorbed.
Factors Affecting Distribution
Rate of blood flow
- greater amounts delivered to organs and tissues with greater blood flow
solubility of drug
- Lipid: readily moves into tissues
- Nonlipid: limited ability to cross cell membranes
Protein binding
- unbound drug = active
effect of protein binding
- limit action
- prolong action
Redistribution
Termination of drug action is normally by biotransformation/excretion, but may also occur as a result of redistribution between various compartments.
Particularly true for lipid-soluble drugs that affect brain and heart.
Barriers to Distribution
specialized capillaries in selected tissues
Placental Membranes
- layer of epithelial cells
blood brain barrier
- endothelial layers of capillaries in brain plus glial sheath
四、Drug Metabolism
Drug Biotransformation
Questions:
- Why is drug biotransformation necessary
- The role of biotransformation in drug Disposition (How)
- Where do drug biotransformation occur
- The enzymes involved in biotransformation (What)
1. The role of biotransformation
Converts pro-drug to active compound
Coverts less active drug to more active drug
Produces toxic compound
Terminates drug actions
2. Biotransformation process-Phase I
Phase 1 reactions usually convert the parent drug to a more polar metabolite by introducing or unmasking a functional group (-OH, -NH2, -SH)
Often these metabolites are inactive, although in some instances activity is only modified or even enhanced
If phase 1 metabolites are sufficiently polar, they may be readily excreted
3. Biotransformation process-Phase II
Phase 2 reactions :An endogenous substrate such as glucuronic acid, sulfuric acid, acetic acid, or an amino acid combines with the functional group to form a highly polar conjugate.
Phase 1 reactions was catalyzed by cytochrome P450 in the hepatic microsome and Phase 2 reactions was catalyzed by some transferases.
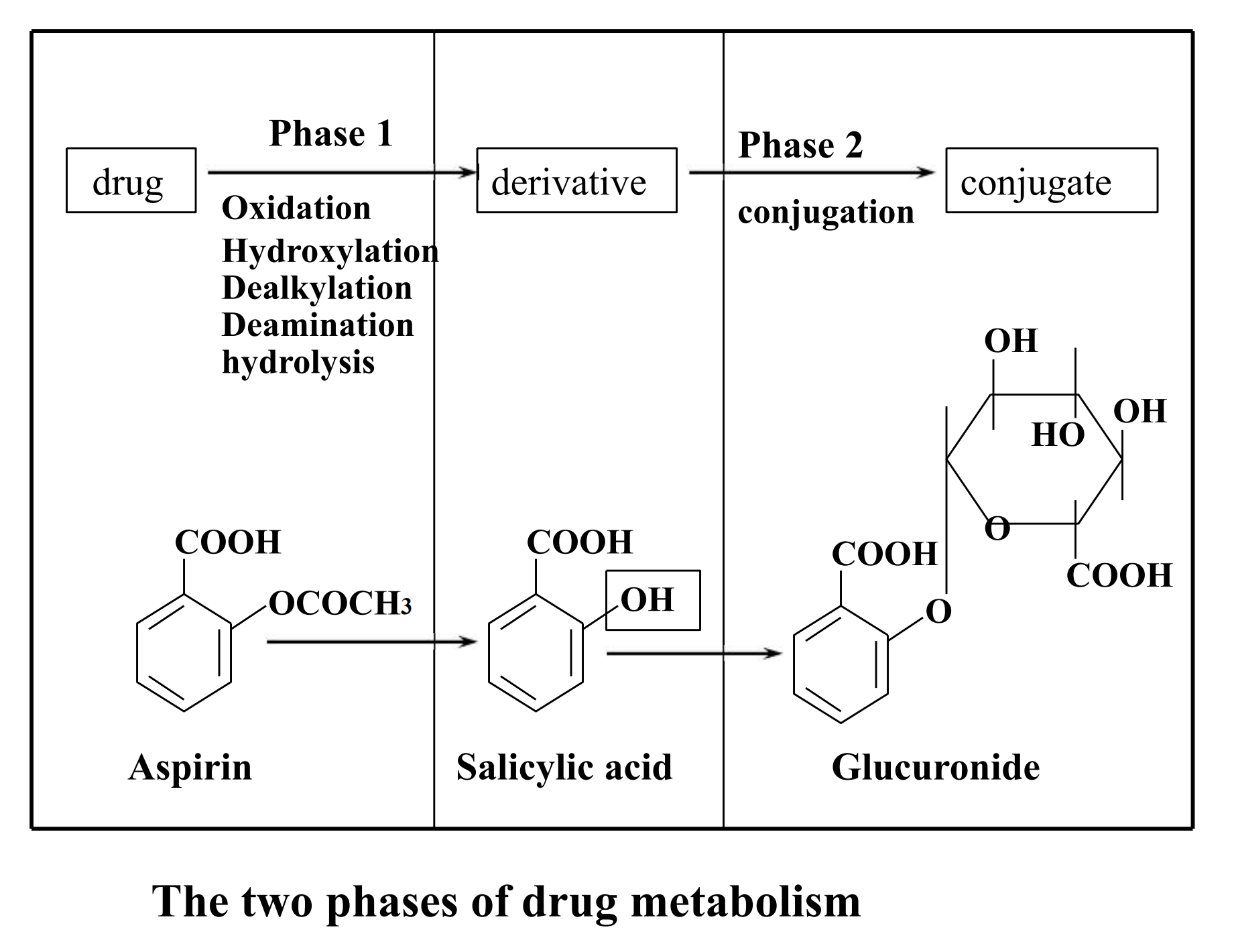
Phase II Conjugation Reactions
Glucuronidation (UDP glucuronosyltransferases)
- Glucuronidation is the most important conjugation reaction
- Enzyme: uridine diphosphate glucuronyl transferases, localized in the microsome
Glutathione (glutathione S-transferases)
Amino acid (glycine, glutamine)
Sulfate (sulfotransferases)
- Sulfation also is an important conjugation reaction
- Enzyme: sulfotransferases, localized in the cytosol
Acetylation (N-acetyltransferases)
Methylation (methyltransferases)
Many conjugation enzymes exhibit polymorphism
Biotransformation of Major Functional Groups
(-OH): oxidation, methylation, glucuronide conjugation, sulfate conjugation
(-COOH): oxidation, glucuronide conjugation, glycine conjugation
(-NH2): deamination (and aldehyde formation), glucuronide conjugation, methylation
Places that Biotransformations Occur
Extrahepatic microsomal enzymes (oxidation, conjugation)
Hepatic microsomal enzymes (oxidation, conjugation)
Hepatic non-microsomal enzymes (acetylation, sulfation,GSH, alcohol/aldehyde dehydrogenase, hydrolysis, ox/red)
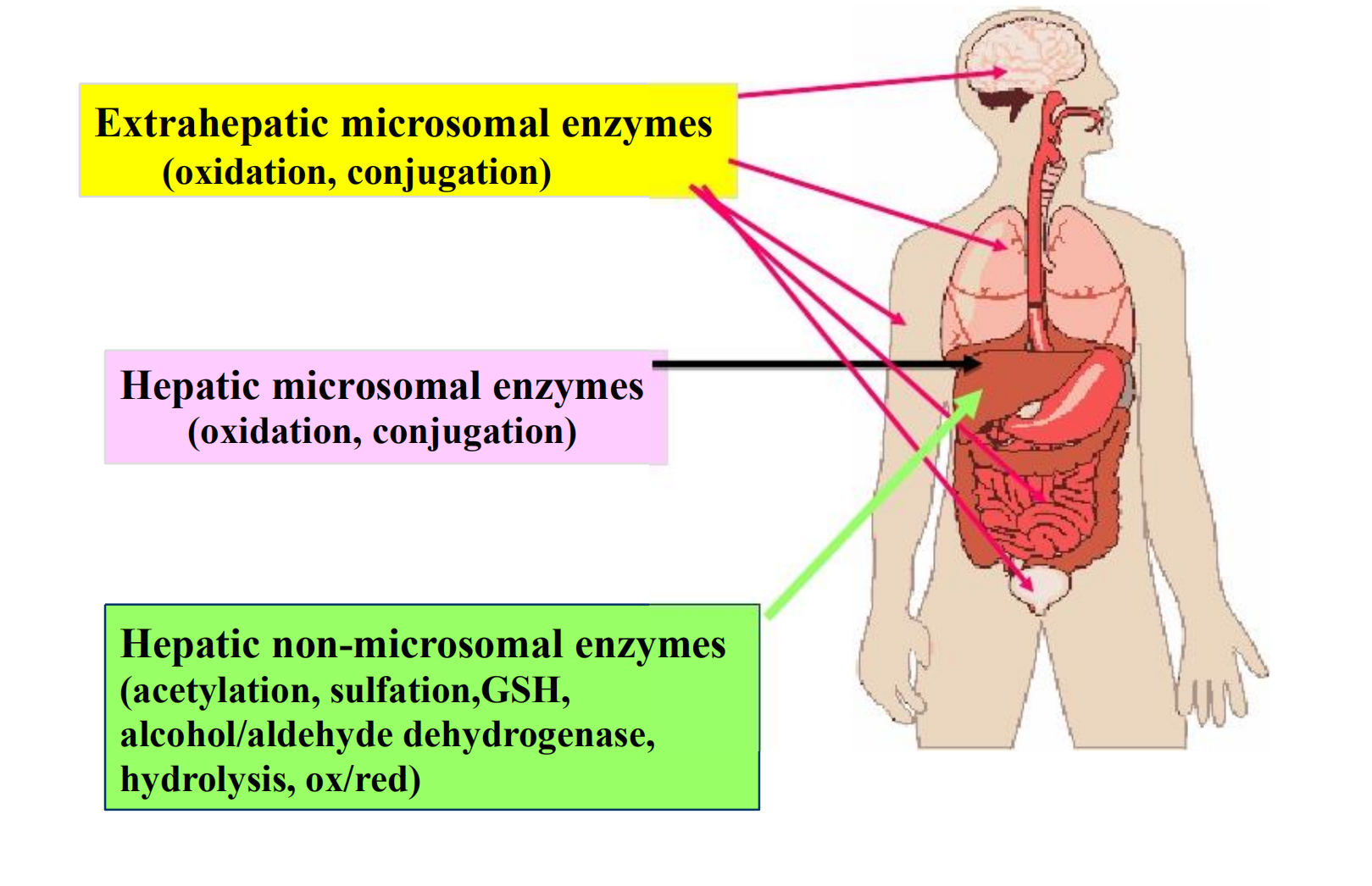
Liver is the principal organ of drug metabolism because of its containing most drug-metabolizing enzymes and its high blood flow
- Gastrointestinal tract, the lungs, the skin and the kidneys
In a cell, the enzymes are localized in the endoplasmic reticulum, mitochondria, cytosol, lysosomes, or even the nuclear envelope or plasma membrane
1. Microsomal Mixed Function Oxidase System & Phase I Reactions
The enzyme systems involved in phase 1 reactions are located primarily in the endoplasmic reticulum.
The ER membranes are isolated by homogenization and fractionation of the cell, they reform into vesicles called microsomes
The smooth microsomes are relatively rich in enzymes responsible for oxidative drug metabolism
Microsomal Mixed Function Oxidase System
This system includes a series of enzymes involved in drug oxidation
One molecule of oxygen is consumed (reduced) per substrate molecule, with one oxygen atom appearing in the product and the other in water
In this oxidation-reduction process, two microsomal enzymes play a key role
- NADPH-cytochrome P450 reductase
- Cytochrome P450
Human Liver P450 Enzymes
Twelve CYP gene families have been identified in humans, and the categories are based upon protein sequence homology
- Families CYP plus arabic numeral (>40% homology of amino acid sequence, eg. CYP1)
- Subfamily 40-55% homology of amino acid sequence; eg. CYP1A
- Subfamily additional arabic numeral when more than 1 subfamily has been identified; eg. CYP1A2
CYP3A4 is very common to the metabolism of many drugs; its presence in the GI tract is responsible for poor oral availability of many drugs
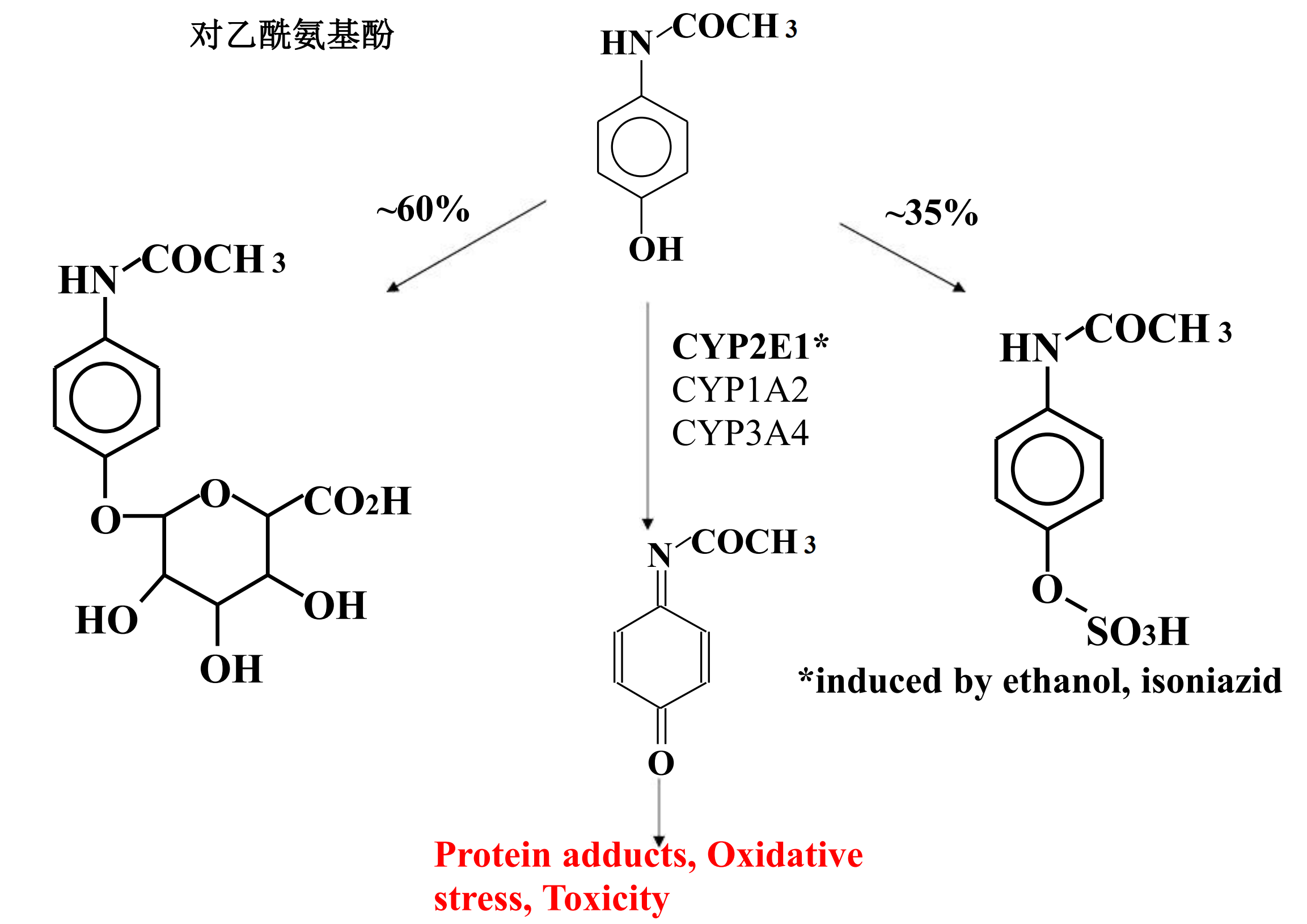
2. Metabolism of Drugs to Toxic Products
Several compounds have been shown to be metabolically transformed to reactive intermediates that are toxic to various organs
- Alternative detoxification mechanisms
- Endogenous detoxifying co-substrates
The number of specific examples of such drug-induced toxicity is expanding rapidly
Acetominophen Metabolism
五、Drug Excretion
movement from tissues to blood to site of removal from body
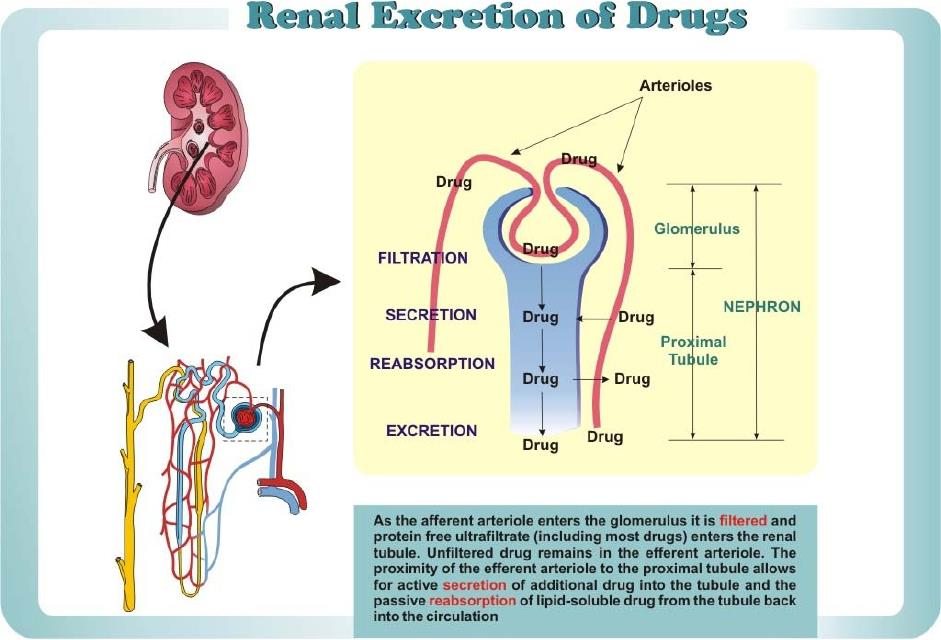
Renal Excretion of Drugs
Filtration (Glomerulus)
Reabsorption Passive Transport (Tubule)
Secretion Active Transport (Tubule)
- Transporter for Organic Acids
- Transporter for Organic Bases
1. Glomerular(肾小球) Filtration
Only unbound drug is filtered.
Plasma Protein Binding of drug prevents filtration: Thyroxine is 99% bound
Molecular Size:
- Albumin (70,000) is not filtered.
- Inulin (5,500) is freely filtered; can be used to estimate GFR.
2. Tubular Reabsorption
Passive Transport:
- pH, concentration, size, lipid solubility, ionization
- acid urine favors reabsorption of weak acid.
- basic urine favors reabsorption of weak base.
Active Transport:
- Uric acid, glucose, amino acids………
3. Tubular Secretion
Active Transport
- Organic Acids (inhibited by probenecid)
- Organic Bases
- No effect of protein binding on this process
4. Secretion Vs. Active Reabsorption
The acid and base transport systems can operate bidirectionally, and at least some drugs are both secreted and actively reabsorbed
However, active transport of most exogenous ions is predominantly secretory.
5. Renal System
lipid soluble drugs are passively reabsorbed
changing pH of urine
- acidic urine =
- alkaline drugs eliminated
- acid drugs reabsorbed
- alkaline urine =
- acid drugs eliminated
- alkaline drugs absorbed
renal disease/ decreased clearance
- affects drug dosage
Hepatic or Biliary Excretion
Some organic acids and bases are actively secreted into the bile
Some drugs are secreted into the bile
- Glucuronides of steroids and morphine are actively secreted
1. Enterohepatic Cycle
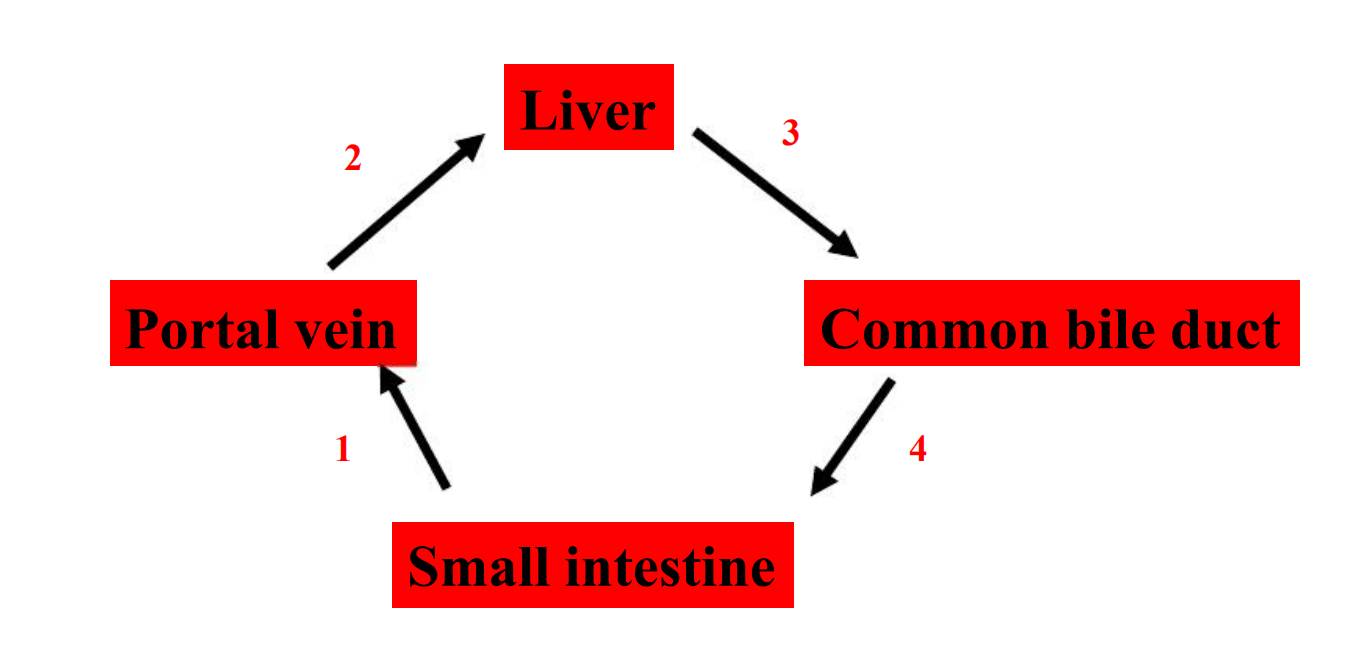
- Prolongs drug half-life
Pulmonary Excretion
Factors:
- Plasma solubility of drug
- Cardiac output
- Respiration
Gaseous Anesthetics
- Rate of pulmonary excretion is proportional to alveolar tension of the gaseous drug and inversely proportional to its plasma solubility
Drug Elimination Kinetics
1. First-order kinetics
A fixed percent/fraction of the drug is eliminated per unit time.
The rate of drug elimination is directly proportionate to plasma concentration
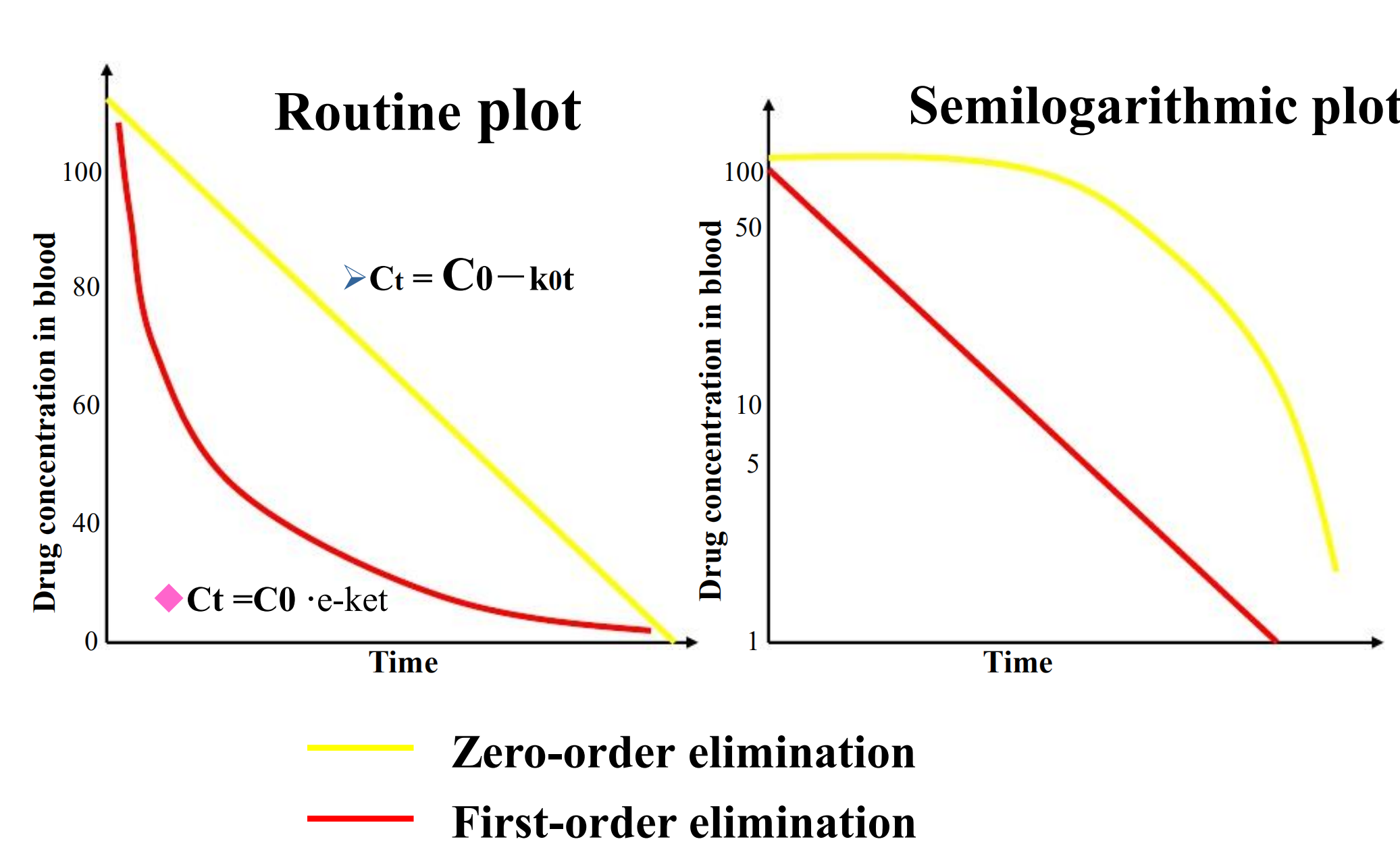
- First-order elimination is also know as constant fraction elimination. Its time-concentration curve presents a curve in a route plot as well as a straight line in a semilogarithmic plot
2. Zero-order kinetics
A fixed amount of the drug is eliminated per unit time
The rate of drug elimination is independent of plasma concentration.
Zero-order elimination is also know as constant quantity elimination. Its time-concentration curve presents a beeline in a route plot as well as a curve in a semilogarithmic plot
3. Michaelis-Menten kinetics
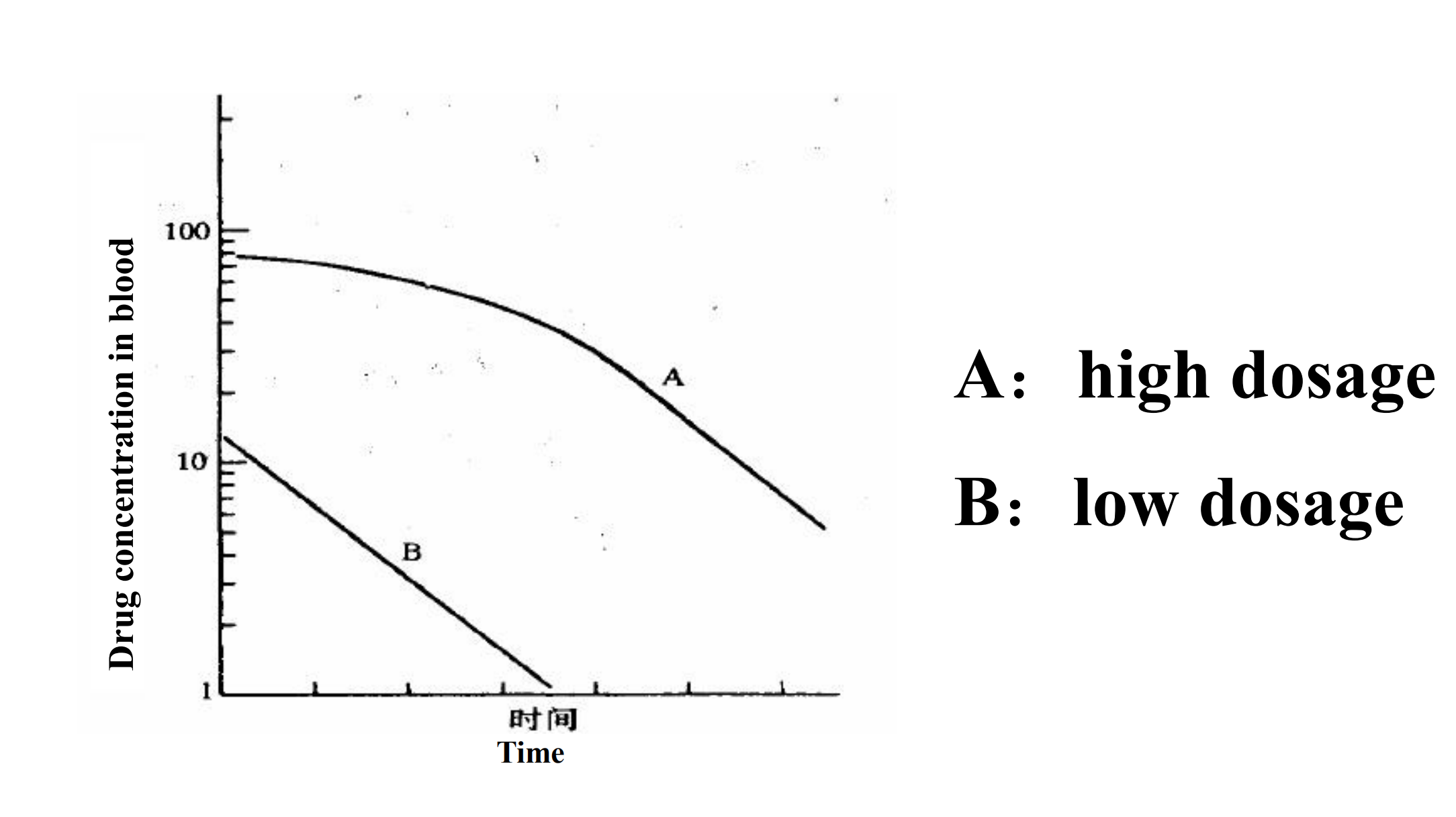
Most drug elimination pathways will become saturated if the dose is high enough.
$$
\frac{dC}{dt} = \frac{V_{max} \cdot C}{K_{m} + C}
$$
- Km represents the concentration at which half of the maximal rate of elimination is reached
- Vmax is equal to the maximal rate of elimination
When Km>>C, the drug elimination capacity is more and more than the amount of drug in body, so the C can be ignored. It can be considered as first-order kinetics
$$
\frac{dC}{dt} = \frac{V_{max} \cdot C}{K_{m}}
$$
When C> > Km , the amount of drug exceed the drug elimination capacity, so Km can be ignored. It can be considered as zero-order kinetics
$$
\frac{dC}{dt} = \frac{V_{max} \cdot C}{C} = V_{max}
$$
Clinical Pharmacokinetics
The four most important parameters are
- Volume of distribution, a measure of the apparent space in the body available to contain the drug
- Clearance, a measure of the body’s efficiency in eliminating drug
- Eliminiation half-life, a measure of the rate of removal of drug from the body
- Bioavailability, the fraction of drug absorbed as such into the systemic circulation
1. Volume of Distribution
$$
v_{d} = \frac{\text{total drug in body (mg)}}{\text{plasma conc. (mg/ml)}}
$$
It is the volume necessary to contain the amount of drug as the same concentration as in the blood
The apparent volume of distribution is a calculated space and does not always conform to any actual anatomic space
Example of Vd
The plasma volume of a 70-kg man ~ 3L, blood volume ~ 5.5L, extracellular fluid volume ~ 12L, and total body water ~ 42L
If 500 μg of digoxin were in his body, Cplasma would be ~ 0.7 ng/ml. Dividing 500 μg by 0.7 ng/ml yields a Vd of 700L, a value 10 times total body volume! Huh?
Digoxin is hydrophobic and distributes preferentially to muscle and fat, leaving very little drug in plasma. The digoxin dose required therapeutically depends on body composition.
2. Clearance
Clearance does not indicate how much drug is removed but, rather, the volume of blood that would have to be completely freed of drug to account for the elimination rate
CL is expressed as volume per unit time.
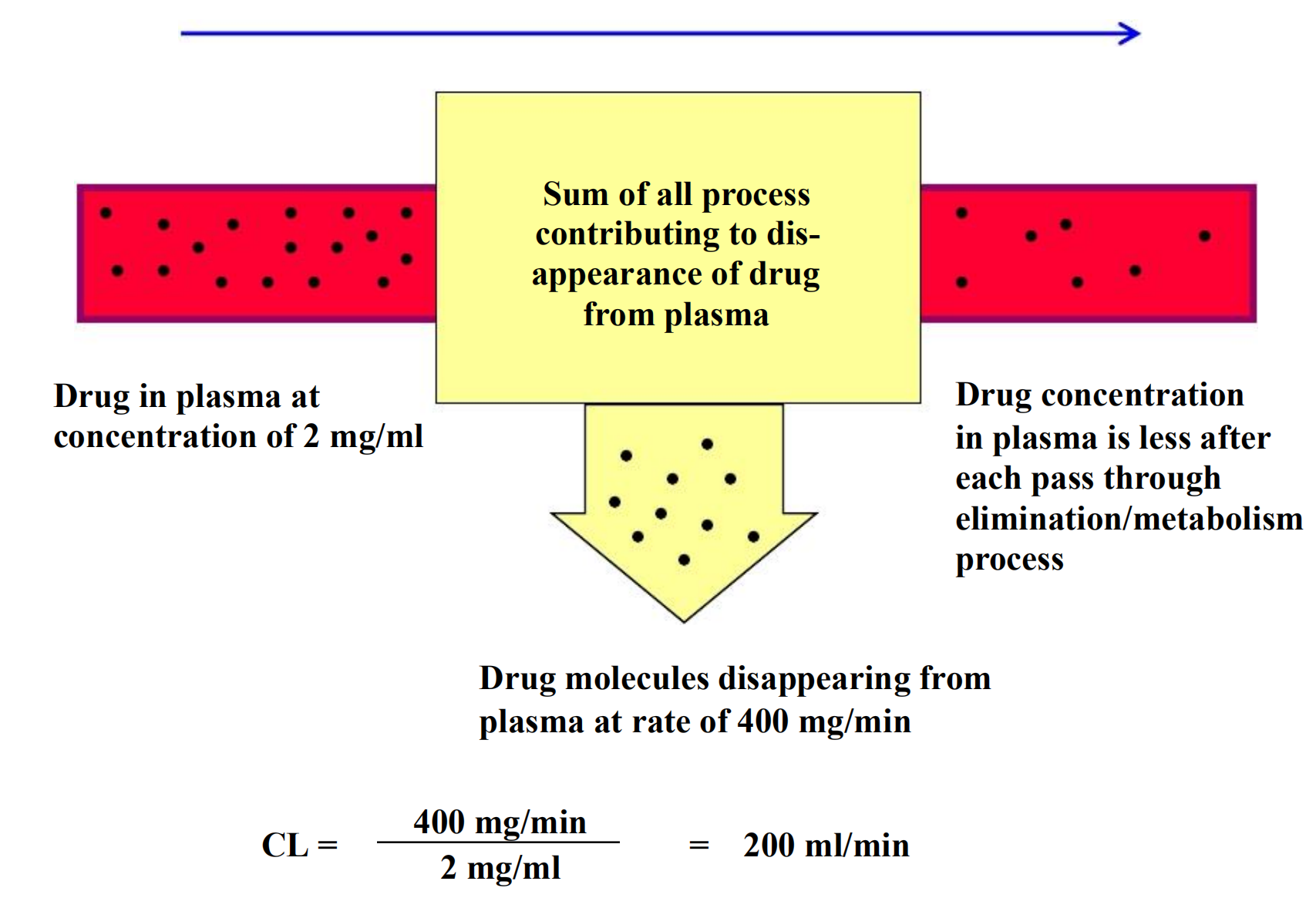
Example: propranolol, CLp = 12 ml/min/kg or 840 ml/min in a 70-kg man.
The drug is cleared almost exclusively by the liver.
Every minute, the liver is able to remove the amount of drug contained in 840 ml of plasma
Clearance of most drugs is constant over a range of concentrations.
This means that elimination is not saturated and its rate is directly proportional to the drug concentration: this is a description of 1st-order elimination.
3. Half Life (t1/2)
Is the time it takes for the plasma concentration or the amount of drug in the body to be reduced by 50%
Indicate the rate of drug elimination in vivo
each half-life: the concentration decreases by half of remaining concentration
Give 100 mg of a drug
- useful in determining drug dosage frequency
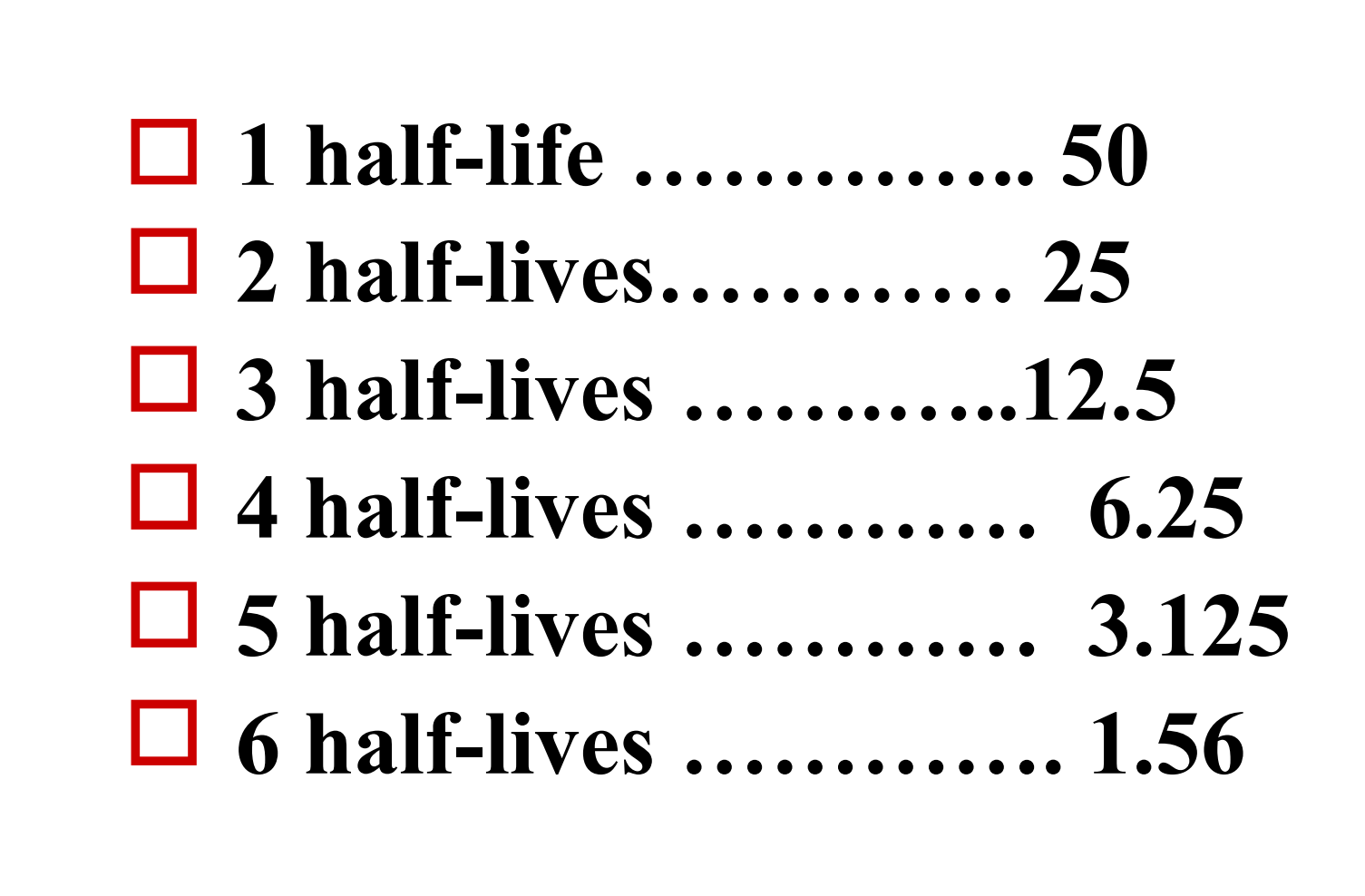
Meanings of t1/2
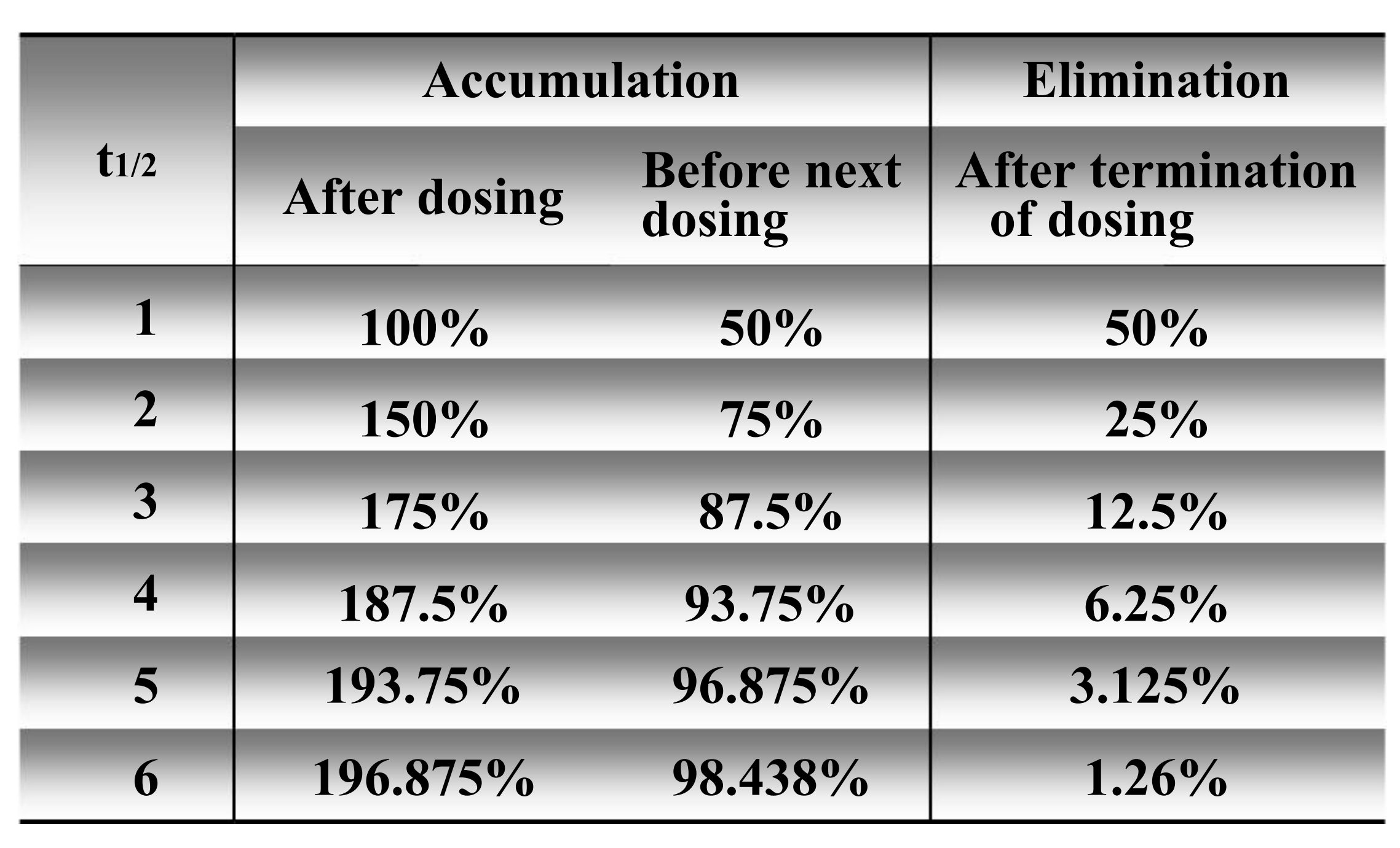
Confirm the interval of administration, generally the interval of administration is one t1/2
Predict the elimination or accumulation of drug in vivo
- As to a single administration, drugs can be eliminated from body after 5 t1/2
- As to continuous administration, drugs concentration can reach steady state concentration after 5 t1/2
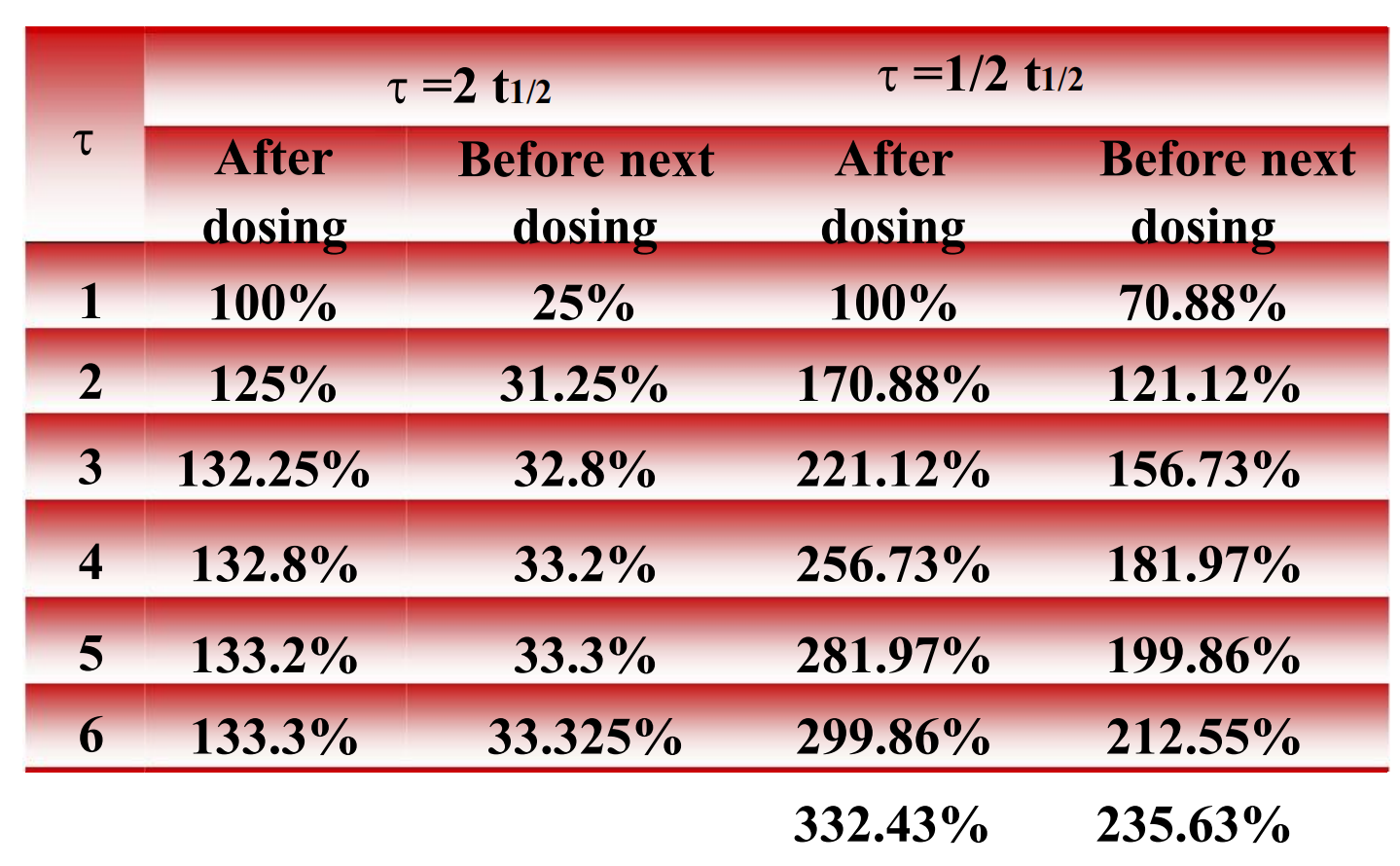
4. The elimination & accumulation of drug
5. Steady-state concentration
The steady state will be achieved when the rate of drug elimination equals the rate of drug administration
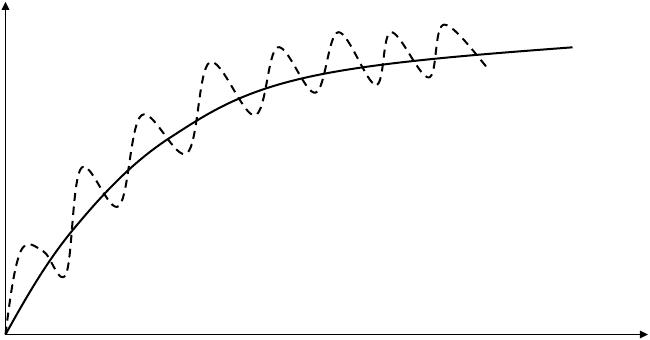
- To the majority of drug eliminated by first-order kinetics, the time achieved to steady state is only determined by the half life
The alteration of the amount of drug in vivo with different dose and same frequency
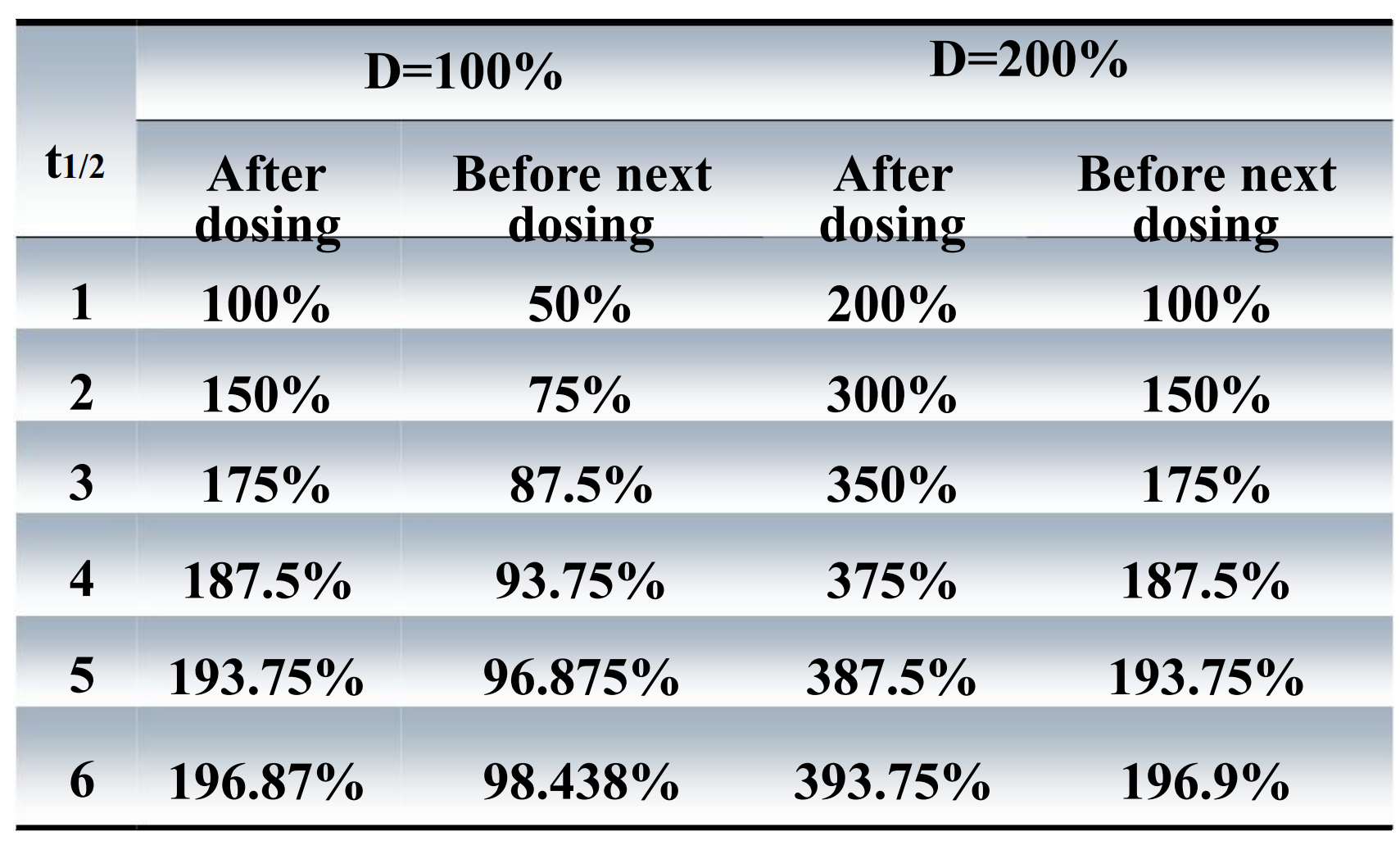
The alteration of the amount of drug in vivo with same dose and different frequency
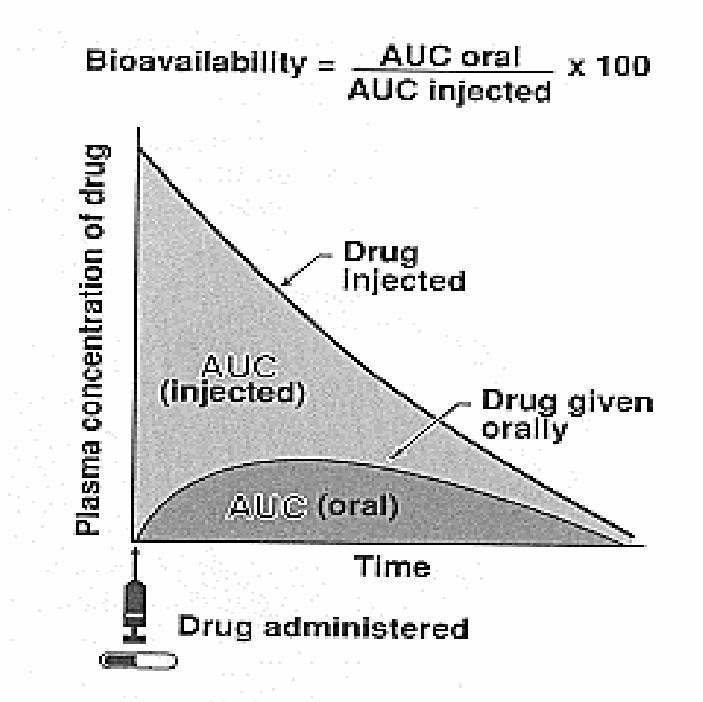
6. Bioavailability (F)
The fraction of unchanged drug reaching the systemic circulation following administration by any route
The area under the blood concentration-time curve (AUC) is a common measure of the extent of bioavailability for a drug.
For an intravenous dose of the drug, bioavailability is assumed to be equal to unity.
AUC (area under the curve)
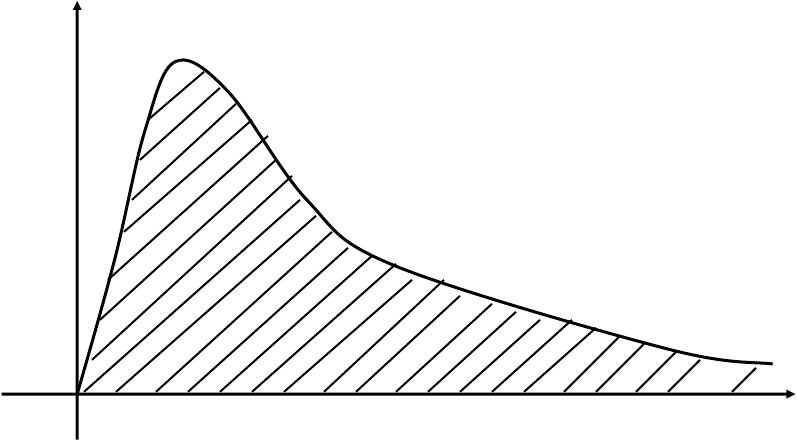
- To multiply concentration by time
Indicates the area under the concentration-time curve
Is an important index to appraise the extent of drugs entering systemic circulation
Unit: (mg/mL)·min, (mg/L)·h, (ng/L)·min, (ng/mL)·h, etc
Bioavailability is used to calculate the PK parameters
- When drugs are extravascular administered, the equations that contain the term dose must include the bioavailability term F, such that the available dose is used
For a drug administered orally, bioavailability may be less than 100% for two main reasons
- Incomplete extent of absorption
- First-pass elimination