L05 Bioinformatics
一、Bioinformatics
Analysis of DNA or protein sequences using computational tools.
Sequencing Techniques
- 454 sequencing technology
- SBS (Sequencing-by-synthesis)
- Illumina sequencing
Definition of Annotation
- Interpreting raw sequence data into useful biological information.
- Addition of information to describe a sequence.
- Information attached to genomic coordinates with start and end point.
- Identification, structural description, characterization of putative protein products and other features in primary genomic sequence.
1. Structural Annotation
Finding genes and other biologically relevant sites thus building up a model of genome as objects with specific locations
2. Functional Annotation
Objects are used in database searches (and experiments); the aim is to attribute biologically relevant information to whole sequence and individual objects
Searching Databases is a kind of functional annotation of sequences
Features that can be annotated in DNA
Genes:
Protein Binding Sites
Alternative Poly-A sites
Alternative Splice sites
Poly-A site
Enhancers: DNA上一小段可与蛋白质(反式作用因子;trans-acting factor)结合的区域,与蛋白质结合之后,基因的转录作用将会加强。增强子可能位于基因上游,也可能位于下游。且不一定接近所要作用的基因,甚至不一定与基因位于同一染色体。这是因为染色质的缠绕结构,使序列上相隔很远的位置也有机会相互接触。
Promoters: 启动子(promoter)是指一段能使特定基因进行转录的脱氧核糖核酸(DNA)序列。启动子可以被RNA聚合酶辨认,并开始转录合成RNA。在核糖核酸(RNA)合成中,启动子可以和调控基因转录的转录因子产生相互作用,控制基因表达(转录)的起始时间和表达的程度,包含核心启动子区域和调控区域,就像“开关”,决定基因的活动,继而控制细胞开始生产哪一种蛋白质。
启动子位于控制基因表达的调控序列中、基因转录起始位点的上游(DNA反义链的5′方向),长约100~1000个碱基对。启动子本身并无编译功能,但它拥有对基因翻译氨基酸的指挥作用,就像一面旗帜,其核心部分是非编码区上游的RNA聚合酶结合位点,指挥聚合酶的合成,这种酶指导RNA的复制合成。因此该段位的启动子发生突变(变异),将对基因的表达有着毁灭性作用。
完全的启动子称为规范序列。3’UTR: 3’非翻译区(英语:three prime untranslated region)简称3’-UTR,是信使核糖核酸(mRNA)中,紧接在编码区之后的序列。mRNA上除了可以翻译成蛋白质的序列外,还有一些不被翻译的序列,如5端(5’)的5′端帽与5′非翻译区(5’-UTR),以及3端(3’)的3′非翻译区(3’-UTR)与多聚腺苷酸尾。3’-UTR上含有AAUAAA的序列,可被特定蛋白识别,以启动多腺苷酸化,即切割mRNA,并于切割位点的3端加上约250个单磷酸腺苷,即多聚腺苷酸尾

Exon and Intron
Non-genic Encoded RNA
- ncRNA(非编码核糖核酸)
- **MicroRNA: ** miRNA通过与目标信使核糖核酸(mRNA)结合,进而抑制转录后的基因表达,在调控基因表达、细胞周期、生物体发育时序等方面起重要作用。
- **Anti-sense RNA: **In genetics, a sense strand, or coding strand, is the segment within double-stranded DNA that runs from 5’ to 3’, and which is complementary to the antisense strand of DNA, or template strand, which runs from 3’ to 5’. The sense strand is the strand of DNA that has the same sequence as the mRNA, which takes the antisense strand as its template during transcription, and eventually undergoes (typically, not always) translation into a protein.
- rRNA
- tRNA
Large Genomic Regions
- Microsatellites: A microsatellite is a tract of repetitive DNA in which certain DNA motifs (ranging in length from one to six or more base pairs) are repeated, typically 5–50 times.(串联重复)
- **Transposons: **A transposable element (TE, transposon, or jumping gene) is a DNA sequence that can change its position within a genome, sometimes creating or reversing mutations and altering the cell’s genetic identity and genome size
- LTRs: (long terminal repeat,长端重复序列)
Long terminal repeats (LTRs) are identical sequences of DNA that repeat hundreds or thousands of times found at either end of retrotransposons or proviral DNA formed by reverse transcription of retroviral RNA. They are used by viruses to insert their genetic material into the host genomes. - heterochromatic knob repeats
Chromosomal Features
- **Telomeres: **端粒(英语:Telomere)是真核生物染色体末端的DNA重复序列,作用是保持染色体的完整性和控制细胞分裂周期。 由于DNA复制的机制,每次染色体复制后,延迟股上的染色体末端必无法被复制。
- Centromere
- Matrix Attachment Regions
Comparative Aspects
- Indels
- SNPs
- Rearrangements
- Duplications
- Synonymous VS. Non-synonymous Substitutions
Gene-finding Programs
GRAIL, MZEF, FGENES, glimmer, GENSCAN, GeneMark, HMMgene….
1. Types of Methods:
Content-based
- codon usage
- substitutions
- composition
- repeats
Site-based
- presence/absence of motif – splice sites
- TF binding sites
- start/stop codons.
Comparative
- make determinations based on sequence homology
2. ORF
In molecular genetics, an open reading frame (ORF) is the part of a reading frame that has the ability to be translated.
An ORF is a continuous stretch of codons that begins with a start codon (usually AUG) and ends at a stop codon (usually UAA, UAG or UGA).
In molecular genetics, an open reading frame (ORF) is the part of a reading frame that has the potential to code for a protein or peptide. An ORF is a continuous stretch of codons that do not contain a stop codon (usually UAA, UAG or UGA).
ORF in cDNA
A cDNA molecule should have only one ORF, meaning that the protein is encoded between one ATG and one stop codon. Other ATGs may be found, including some in the 3’ UTR , but there will be very few ORFs (only one?) per mRNA/cDNA.
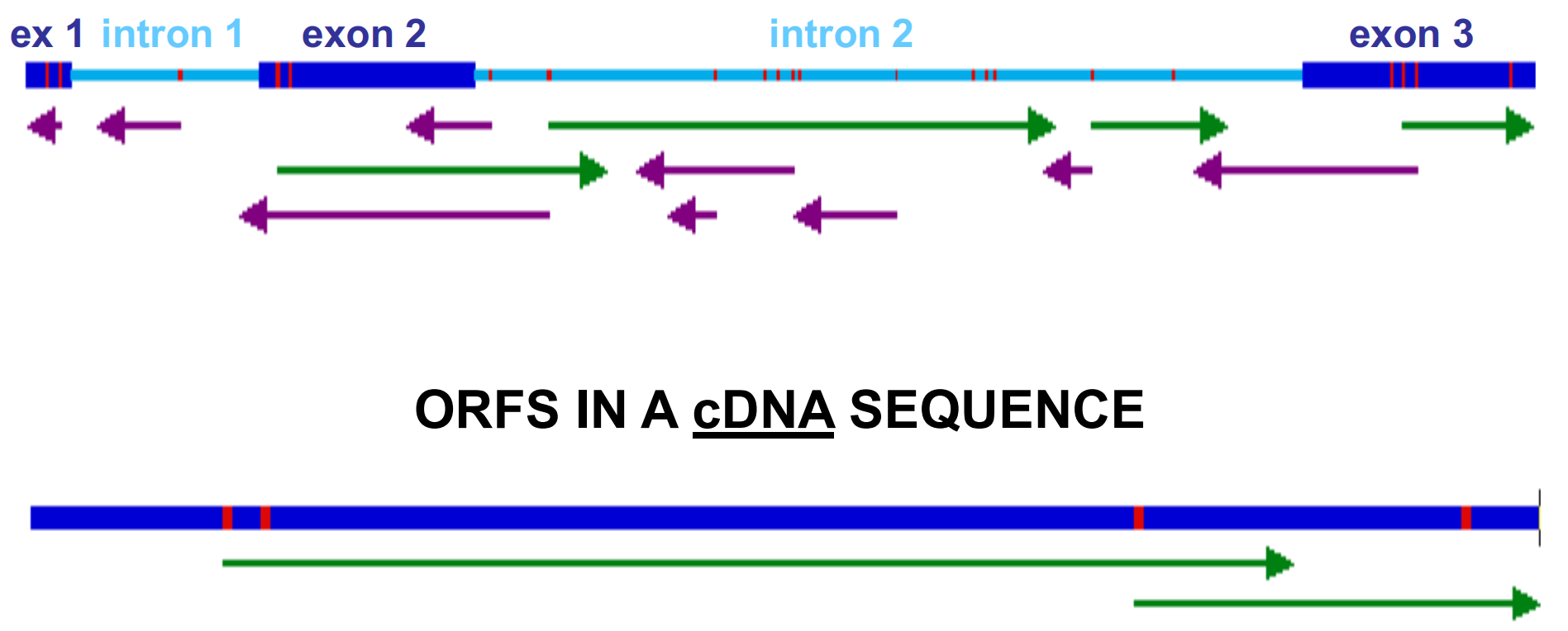
3. Steps to Find Gene
The first step in gene finding is to identify all of the ORFs (open reading frames). These start with ATG, ends with in-frame TAA, TGA, or TAG.
Only a fraction of these will include the entire gene, but many will be exons.
As a general rule, for a given gene, there is only one ATG that is used as a translational start. However, there may be internal ATGs that are just normal methionine codons.
Searching Databases
Functional annotation of sequences: Searching Databases
实验室内的实验很花时间,因此想要得到某些信息的时候,搜索database很方便快捷,同时也很cheap
- Databases can indicate known function of related sequences (predict function based on similarities in structure)
- Database homology searches can annotate large numbers of unknown sequences (e.g. whole genomes or sets of ESTs).
1. BLAST – The king of search tools
Basic Local Alignment Search Tool – most commonly used program due to large size of databases.
Heuristic algorithm to accelerate searches; aligns small bits of DNA and looks for the best overall match between the “query” sequence and an entry in the database.
The BLAST programs are very fast because they only calculate optimal alignment scores for most closely related sequences.
BLAST compares short segments (words) of the query and database to find alignments that exceed a particular score.
2. Different Favors of BLAST
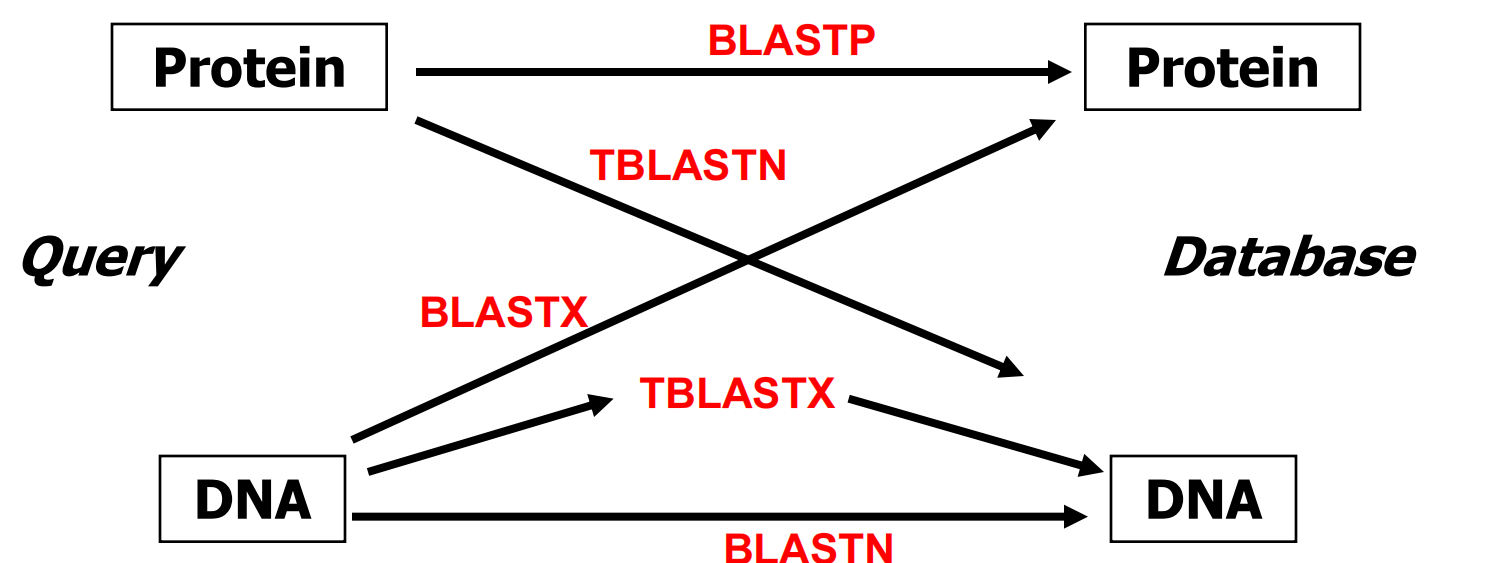
All comparisons except BLASTN are carried out as protein/protein comparisons.
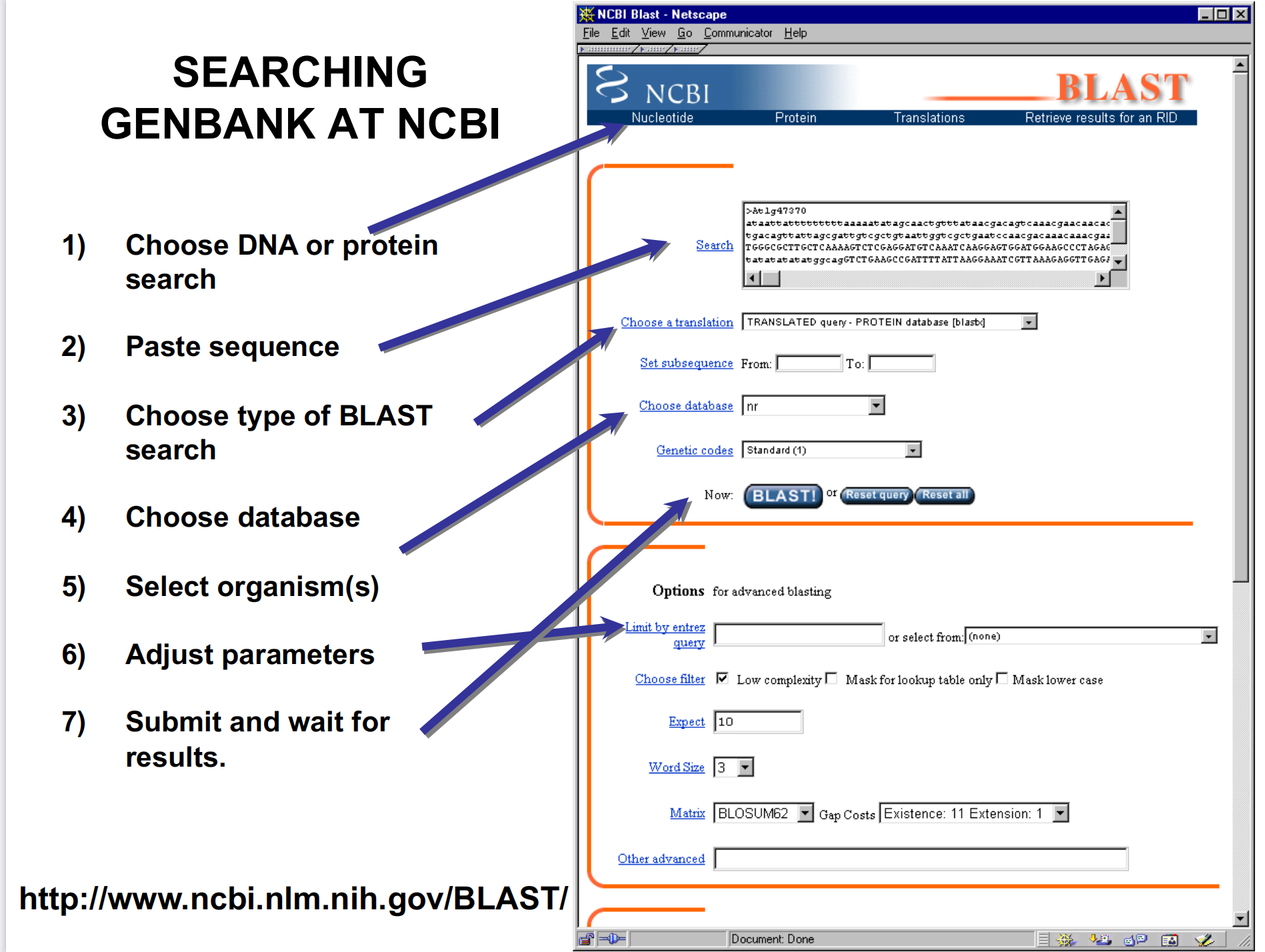
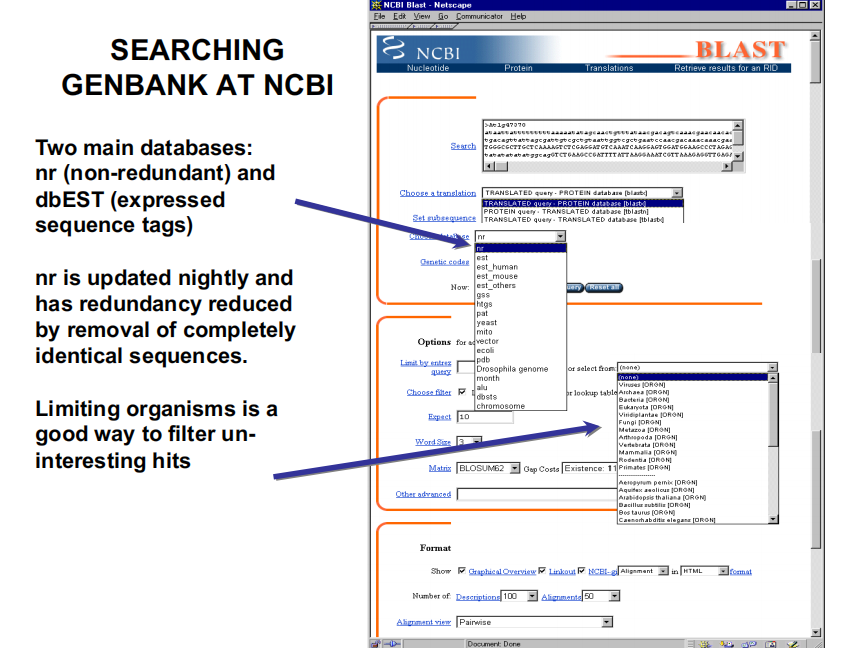
3. How to USE – Searching Genbank At NCBI
4. Schematic of BLASTN results
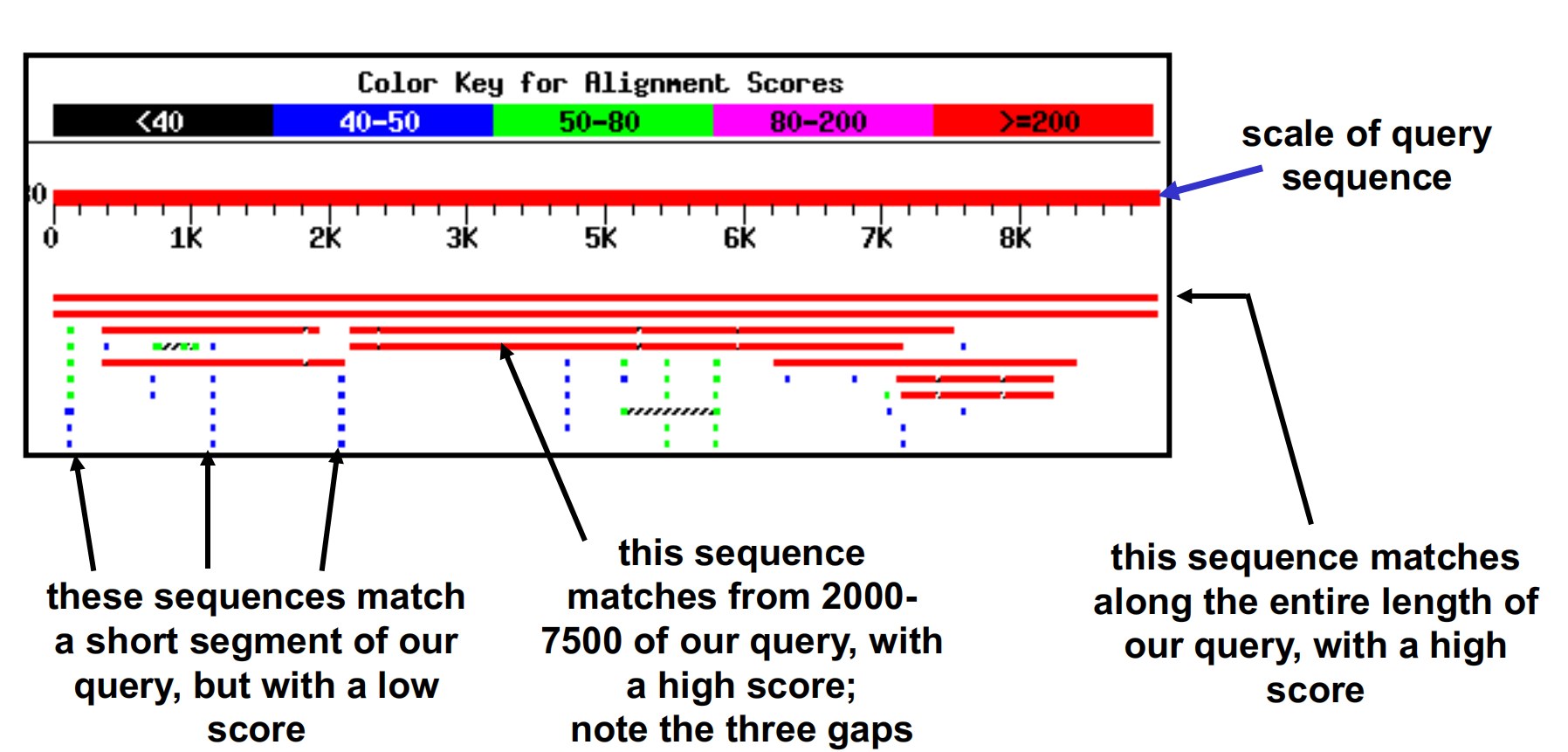

5. How to read the BLAST Result
Bit score – normalized from the raw score, and comparable between alignments. The raw score “S” of an alignment is usually calculated by summing the scores for each letter-to-letter and letter-to-null position in the alignment.
P-value: The number of random local alignment scores equal or greater than S is described by the Poisson distribution. This P value is associated with the score S.
Highly significant scores have P values close to zero.

The Expect value (E) is a parameter describing the number of hits “expected” by chance when searching a database of a particular size. Decreases exponentially with the score. The E value describes the random background noise that exists for matches between sequences.
Sequence Alignment 序列比对
DNA and Protein sequence homology can be used to infer evolutionary relations.
二、The characteristic used for bioinformatic
Motifs
Protein motifs are short conserved segments that have a particular function.
The presence of certain motifs can be used to predict basic characteristics of a protein that otherwise you may know nothing about.
Similarities in protein sequence motifs indicate related functions

Homologous
DNA and protein homology can be used to infer evolutionary relationships
Coding sequences are more highly conserved than non-coding sequences
Natural selection acts on the protein because that is where the function is., but the result of the selection is stored in the DNA. If a protein is advantageous, it is the gene that is conserved.
As shown below, there is little conservation in introns, because there are no or few known functions in introns that selection can act on.
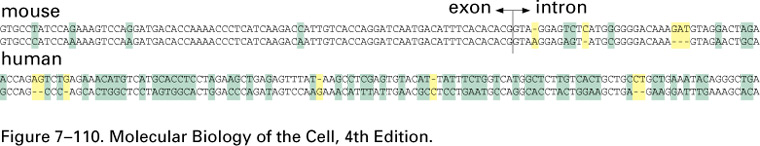
Homologous proteins resulted from gene duplication, not convergence. Related organisms show sequence homology at the level of DNA and Protein
Mutations occur randomly and at a very low frequency along the gene due to mistakes in replication of DNA during cell division
Proteins with similar sequences will have similar structure and/or function (broadly).
- True biological function needs to be determined experimentally!
三、Tools For Analyzing Gene Expression
Transient/Stable expression arrays
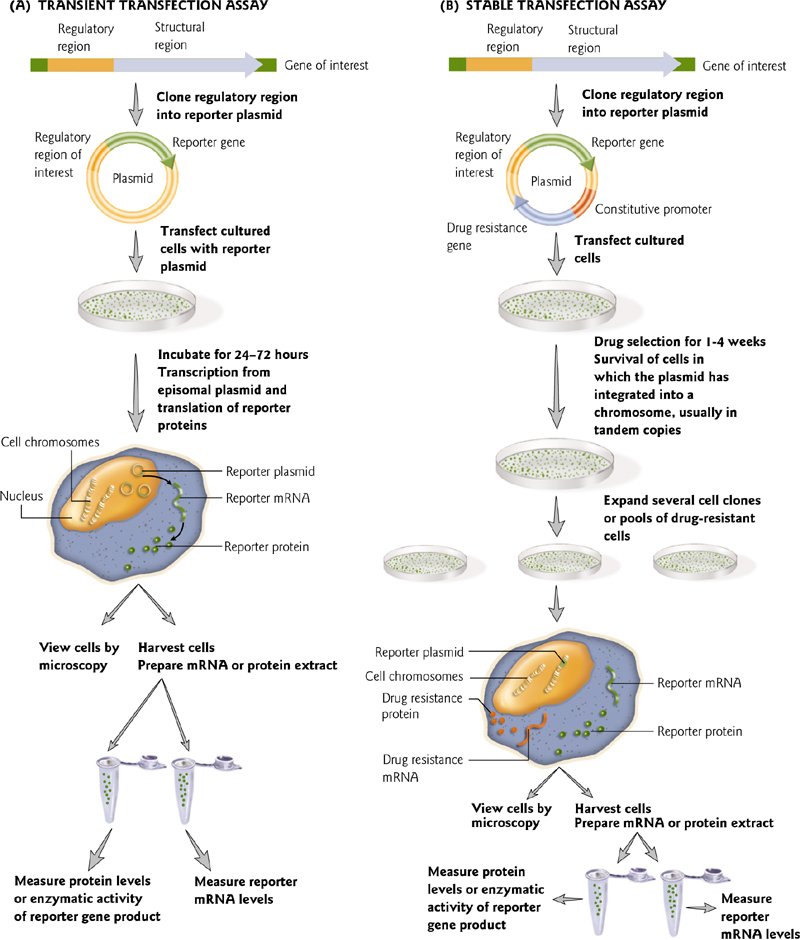
Reporter gene 报告基因
Reporter genes encode proteins or enzymatic functions not typically found in a cell.
报告基因(英语:reporter gene,又称报导基因)是一个分子生物学概念,它是指一类在细胞、组织/器官或个体处于特定情况下会表达并使得他们产生易于检测、且实验材料原本不会产生的性状的基因。较常用的报告基因有绿色萤光蛋白基因(GFP,Green Fluorescent Protein)、β-半乳糖苷酶基因(LacZ等)等。
Example:
- B-galactosidase (color) Luciferase from fireflies Chloramphenicol acetyltransferase (CAT)
- CAT activity can be measured by acetylation of chloramphenicol and separation by thin layer chromatography.
- GFP
- β-glucuronidase (GUS) gene
- Leaf vascular patterning in Arabidopsis shown via expression of the β-glucuronidase (GUS) gene in Arabidopsis leaves
1. GFP and GFP-like
GFP is one of the most commonly used reporter genes.
GFP contains a fluorescent compound (fluorophore). Different mutations result in altered spectra of fluorescence. Excitation takes place at one wavelength and fluorescence at another.
GFP can be used for GFP expression plasmid or GFP expression protein.
Some GFP-like proteins can attach to specific RNA, then make the RNA molecular emit light.
2. Fluorescence Dye (In Situ Hybridization)
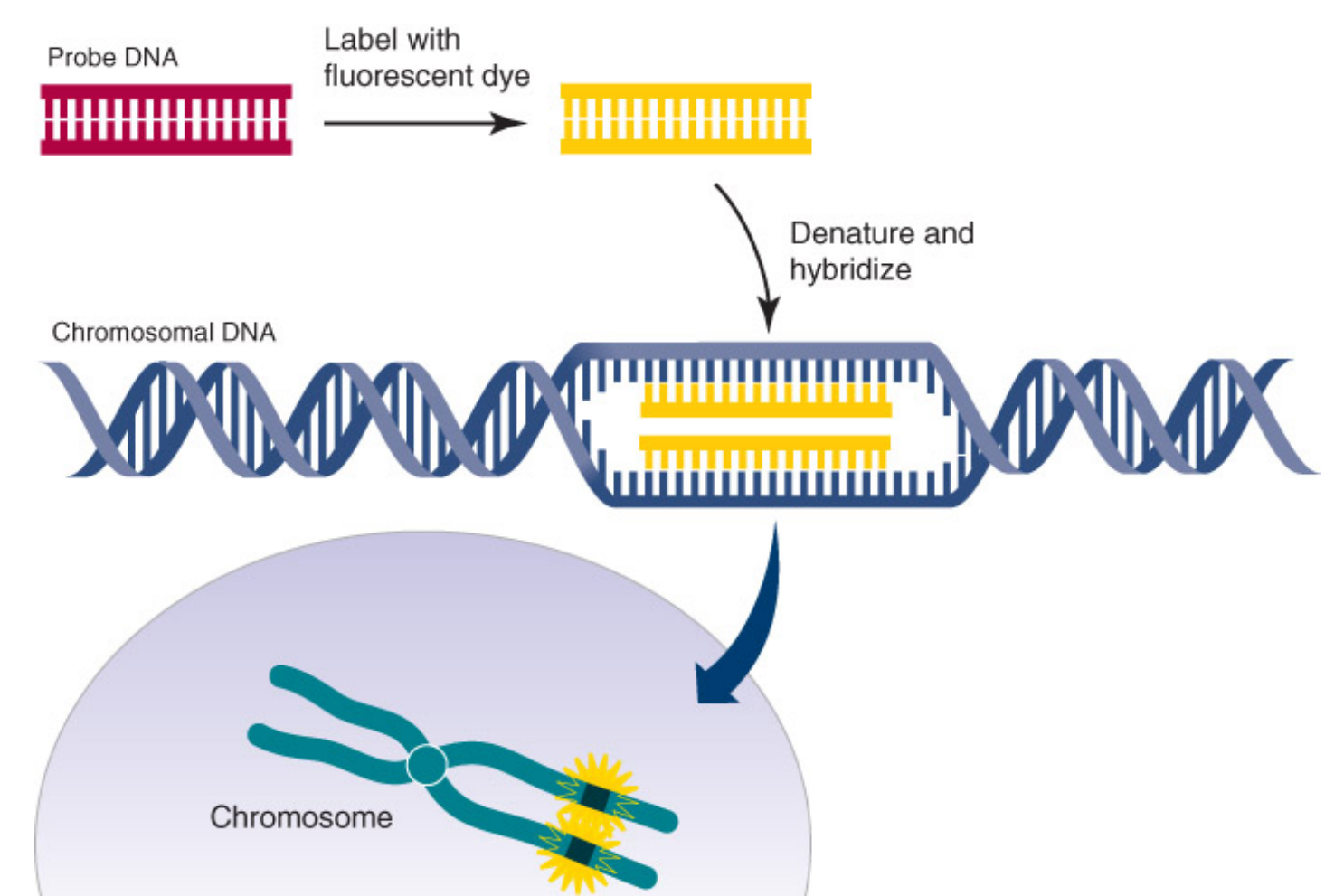
The fluorescence dye can be used to DNA and RNA
Co-translation Assay
Tests for the ability of a DNA-binding protein to activate a reporter gene regulated by the control region of interest.
Hybridization probe 杂交探针
杂交探针(英语:Hybridization probe)是一小段单链DNA片段(十几到几百个碱基),用于检测与其互补的核酸序列。双链DNA加热变性成为单链,随后用放射性同位素(通常用32P)、荧光染料或者酶(如辣根过氧化物酶)标记成为探针。32P通常被掺入组成DNA的四种核苷酸之一的磷酸基团中,而萤光染料和酶与核酸序列以共价键相连。
当将探针与样品杂交时,探针和与其互补的核酸(DNA或RNA)序列通过氢键紧密相连,随后,未被杂交的多余探针被洗去。最后,根据探针的种类,可进行放射自显影、荧光显微镜、酶联放大等方法来判断样品中是否含有,或者何位置含有被测序列(即与探针互补的序列)。
Purification and Detect Protein Tags
Microscopy
Protein Gel Electrophoresis
Polyclonal Antibody
Analyze at RNA Level
- Northern blots for size and abundance.
- In situ hybridizations for cellular localization.
- RNase protection assay – an RNA probe is hybridized to total RNA, protecting the target from digestion by RNase.
- RT-PCR for rapid abundance data.
四、Methods to Analyze Protein Expression
- Western blots (immunoblotting) for size and specificity.
- In situ hybridization for localization using secondary, labeled antibodies.
- Enzyme-linked immunosorbent assay (ELISA, 酶联免疫吸附试验) – a colorimetric assay for protein levels.
五、Inhabitance or Interference to Gene Expression
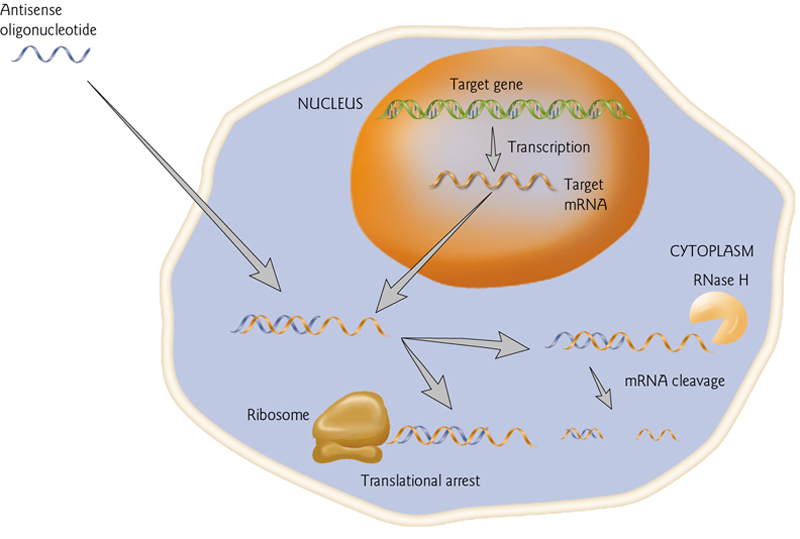
Antisense-oligo(反义寡核苷酸) Mediated Inhibition of Gene Expression
Antisense oligos complementary to mRNAs can target mRNA for degradation or block translation.
RNA Interference
Processing of dsRNA from viral or exogenous(外源的) sources.
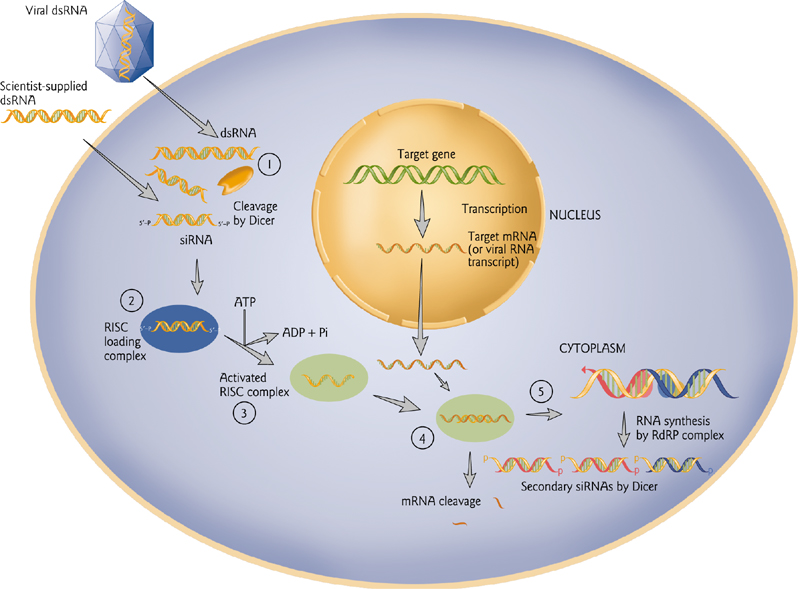
Artificial microRNA can be used to down-regulate Genes or virus
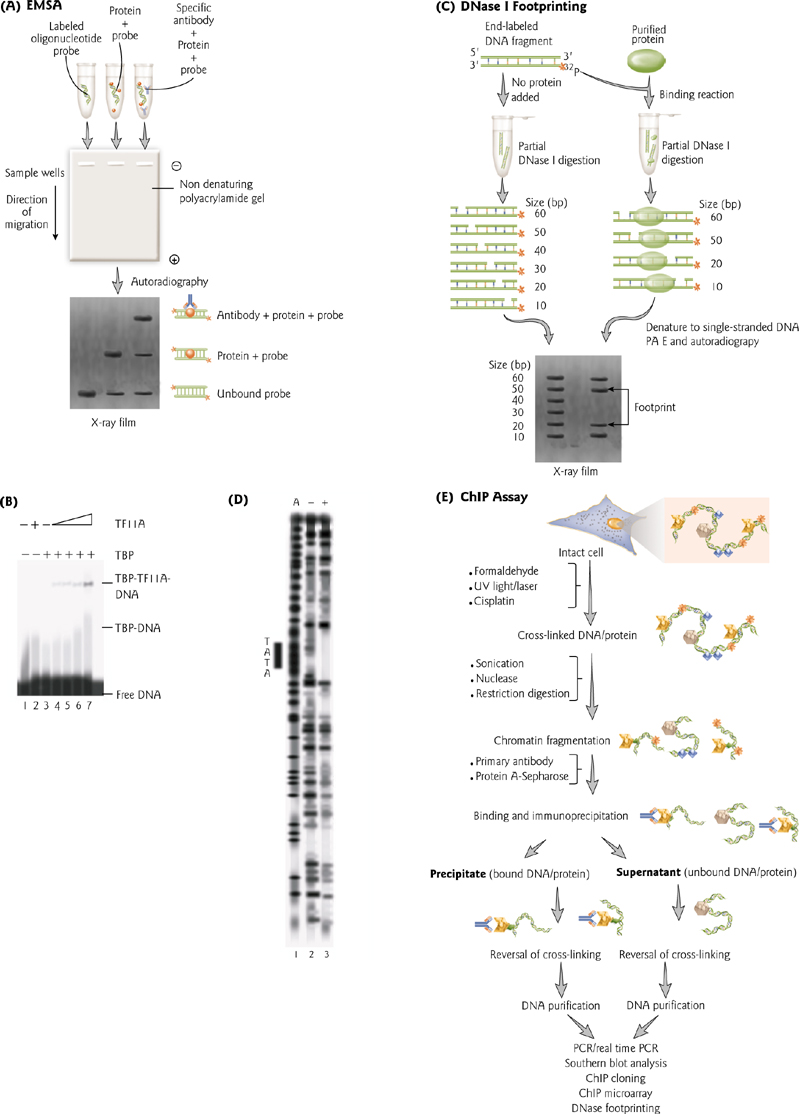
六、Methods for analyzing DNA-Protein Interactions
- Electrophoretic mobility shift assay. DNA is labelled with 32P. If bound by protein, it causes shift in size.
- DNase footprinting. Protein binding “protects” DNA from digestion by Dnase.
七、Methods for analyzing Protein-Protein Interactions
- GST pull-down assay to see which proteins interact with a GST-tagged protein of interest.
- Yeast two-hybrid. Interaction of two proteins activates expression of reporter.
- Co-IP to see if one protein can precipitate another through a direct interaction.
- Fluorescence resonance energy transfer. Energy is only transferred when two tagged proteins are within 10 nm.
八、Structural Analyze for Protein X-ray crystallography
- X-ray crystallography uses diffraction to determine crystal structures
- AFM determines the properties of the surface of a material.
九、Analysis of Gene Expression In Plants
Assessing Transcript Abundance From Extracted RNA(评估提取的RNA的转录丰度)
Poly(A)+RNA can be separated from other RNAs by FRactionation on An Oligo(dT) Column
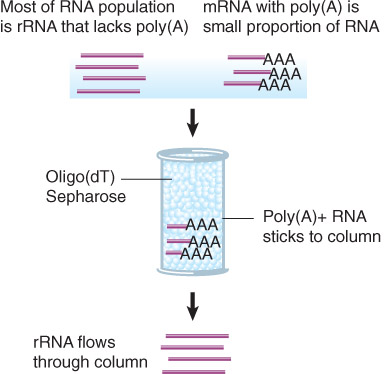
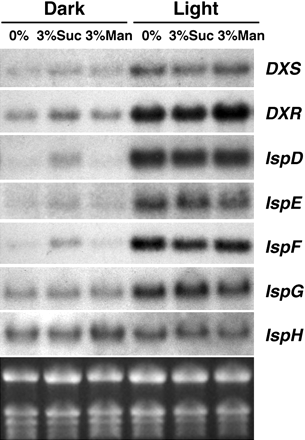
Northern Blots
北方墨点法(英语:Northern blot)是分子生物学、生物化学中常用的实验方法,通过探测样品中的RNA(或隔离的mRNA)来研究基因表现。
利用探针侦测由凝胶电泳分离出来的RNA片段,寻找含有特定串行的RNA片段。与南方墨点法相似,RNA样品经过凝胶电泳后会按照大小分离,然后将样品从凝胶转印到膜上,并用与目标串行互补的标记的探针来探测。实验结果可以根据所用探针的不同以多种方式来观察,但大多数都显示的是样品中被探测的RNA条带的相对位置,也就是分子大小;而条带的强度则与样品中目标RNA的含量相关。这一方法可以测量目标RNA在不同样品中的情况,因此已经被普遍用于研究特定基因在生物体中表现的时刻和表现量,也是这类研究中最基本的手段。
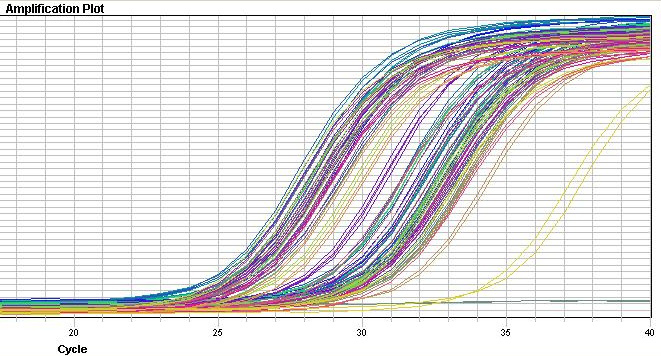
Real-Time PCR
PCR used to measure amplification curve of individual genes
Amplification performed with dye-labeled primers to enable comparisons within a tube
Microarrays
A microarray is a multiplex lab-on-a-chip. It is a 2D array on a solid substrate (usually a glass slide or silicon thin-film cell) that assays large amounts of biological material using high-throughput screening miniaturized, multiplexed and parallel processing and detection methods.
Types:
- cDNA microarray
- High-density oligonucleotide microarrays
Sequencing Methods
1. ESTs
Sequencing of mRNAs, converted to cDNA
“Digital” measurements because counting frequency of transcripts instead of “analog” measurements of hybridization intensity
EST sequences were used as starting material for microarray design
Cloned cDNAs used for EST sequencing were spotted on arrays
2. SAGE
The first “digital” transcriptome sequencing method to use specifically captured short “tags” of cDNAs.
Allowed sequencing of many tags per Sanger sequencing read.
Tags mapped back to longer ESTs or genomic sequences to measure abundances
3. DGE (Digital Gene Expression)
Like SAGE, “digital gene expression” methods capture short tags from specific locations at the 3’ end of end of the transcript.
Longer tags than most SAGE, and optimal for Illumina sequencing
These tags are useful for quantification by counting, or for measuring specific types of transcripts.
4. RNA-SEQ
The most popular off all methods at the present time 一 fragmented mRNA/cDNA, deeply sequenced by next-gen methods.
5. PARE
Data analysis of sequencing data is more “tricky” than microarray data due to the requirement to match reads back to the gene or genome, then add them up and normalize them.
Experimental Design
An important aspect of both array and sequencing experiments
Considerations:
Scientific
Aim of the experiment.
Specific questions to be answered and how they are prioritized.
How will the experiments answer the posed questions?
Practical (logistic)
Types of mRNA samples: reference, control, treatment 1 (T1), and so on.
Amount of material available: count the amount of mRNA involved in one
channel of one hybridization as one unit.
Number of slides available for the experiment.
Other factors
The experimental process before hybridization: sample isolation, mRNA
extraction, amplification and labelling.
Controls planned: positive, negative, ratio, and so on.
Verification method: northern blot, reverse transcriptase (RT)-PCR, in situ hybridization, and so on.