L5 DNA sequencing
First generation sequencing
- Maxam & Gilbert Sequencing
- Sanger Sequencing
Next-generation sequencing/high-through put sequencing
- Illumina (Solexa) sequencing
- Roche 454 sequencing
- Ion torrent: Proton / PGM sequencing
- SOLiD sequencing
Third/Fourth generation sequencing
- PACBIO sequencing
- Nanopore sequencing
一、Maxam-Gilbert Sequencing
It is a method by which the sequence of a DNA fragment is identified by using chemicals, which means cut DNA at specific points.
- Also called Chemical degradation method of DNA sequencing
Procedure
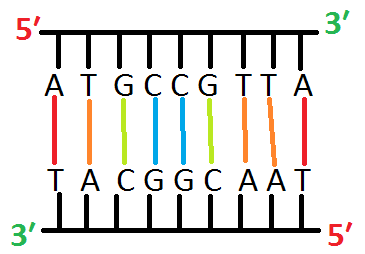
- E.g it is a fragment of double stranded DNA, and you do not know its sequence
Step:1, Alkaline denaturation of dsDNA
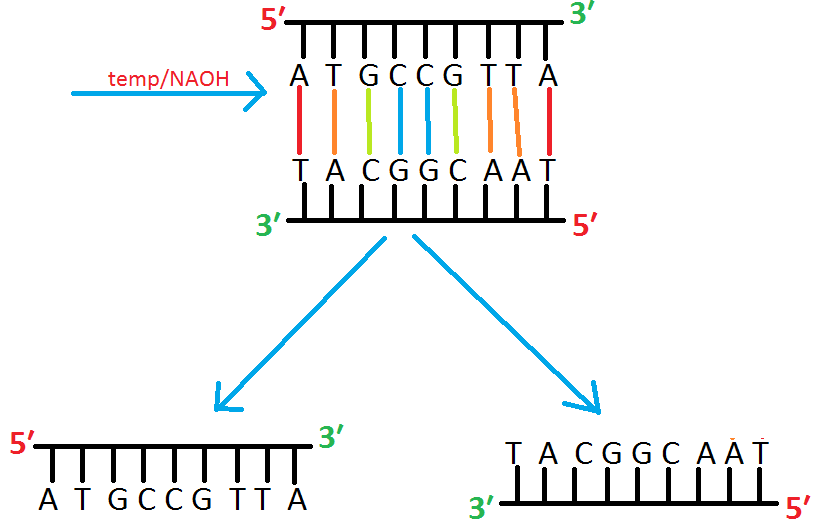
- At first, the double stranded fragment is separated into two single strands by applying high Temperature or high PH.
Step :2 Separate two strand on gel
Run the single stranded fragments on gel.
- As lighter fragment band will move further than the heavy fragment band
How will we know which one is the lighter band?
- The band having larger number of purines (A,G) would be heavier.
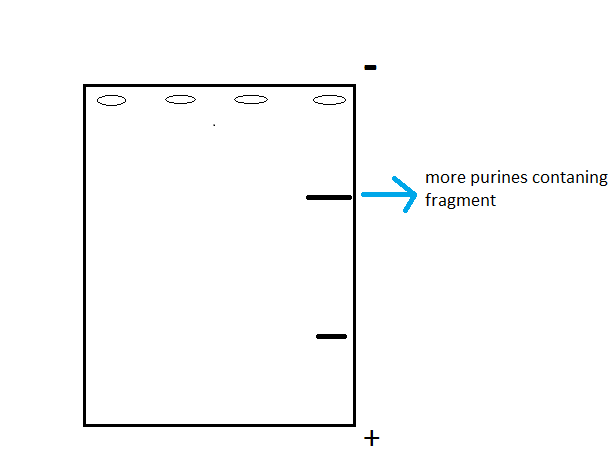
- Fig: Single stranded DNA fragments in Gel Electrophoresis gel.
Step :3, Radioactive end labeling of ssDNA
Take one of fragment band from the gel

Remove the Phosphate at 5′ end and incorporate radioactive 32-PO4 enzymatically.
Step 4: Chemical Degradation
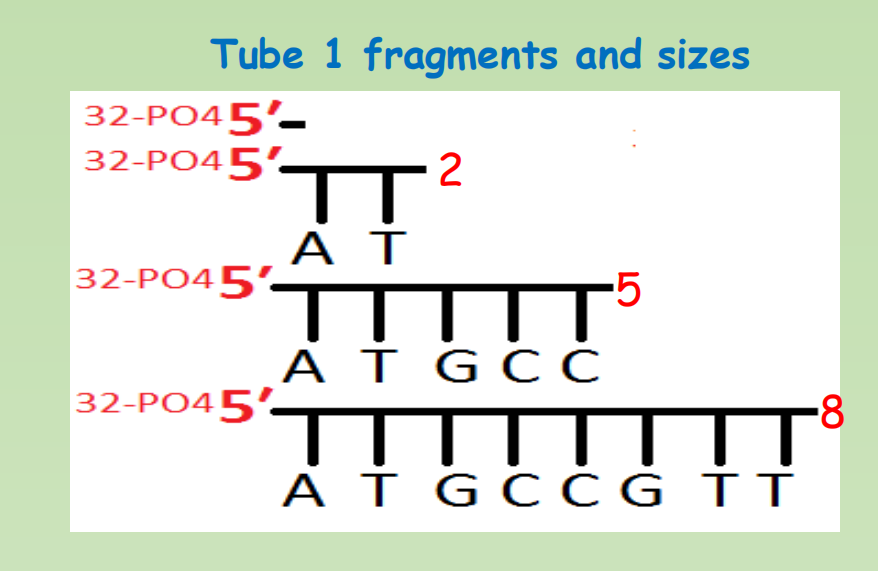
Now let’s put isolated and end-labelled ssDNA into four tubes
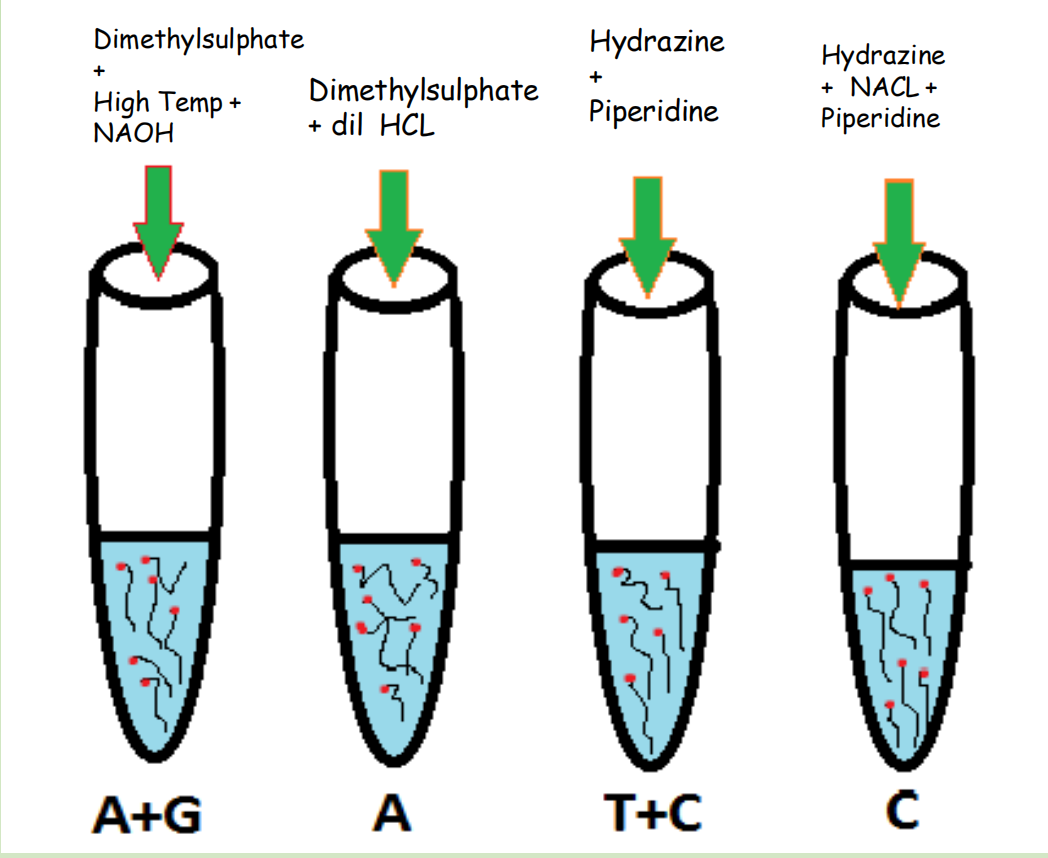
- Tube 1 : Increase Temperature and PH(by adding NAOH), that would cause fragments to break down. Dimethyl sulfate will be added that would make cuts at Adenine and Guanine positions.
- Tube 2: Dimethyl sulfate and dilute HCL will be added that would cuts the fragment at Adenine position
- Tube 3: Reagents Hydrazine and Piperidine are added that would cuts the fragment at position Cytocine and Thymine
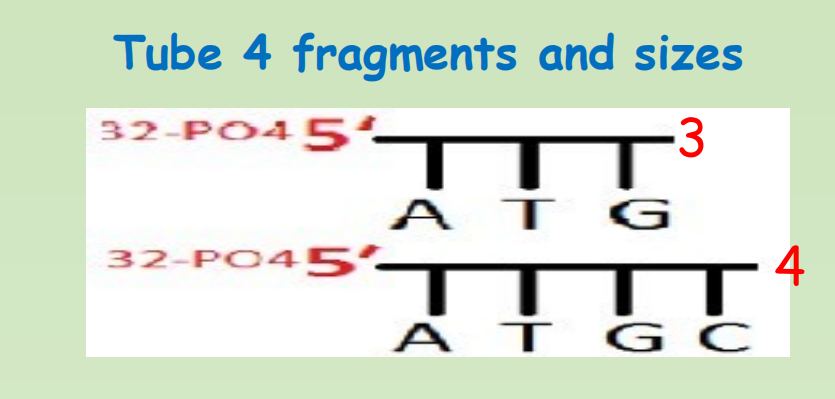
- Tube 4: In the last tube, Hydrazine, Piperdine and NACL is added that would cuts the fragment at Cytocine position
After chemical degradation, we would get following radioactively labelled fragments from each tube


Step 5: Gel electrophoresis
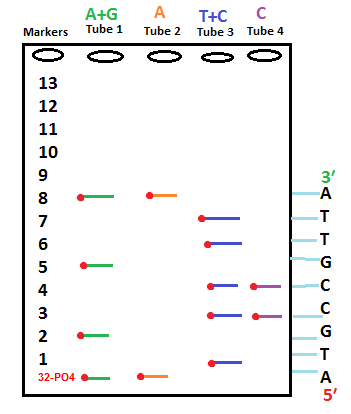
- All of the fragments from each four tubes are pour in Gel.
- Four wells will be make on Gel with each tube of the samples running into one well.
- Fragments would separate on Gel according to size.
- After placing X ray film on top of gel, radioactive labelled fragments would emit a band at their position
Advantages
- Directly read purified DNA.
- Used sequence heterogenous DNA as well as Homopolymeric sequences.
- used to analyze DNA-Protein interaction.
- Used to analyze Epigenetic modification and nucleic acid structure.
Disadvantages
- Use of toxic chemicals and extensive use of radioactive isotopes. highly poisonous and unstable
- Cannot read more than 500bp
- Setup is quite complex. (Chemical digestion, Gel running, radioisotope, time consuming)
- It is difficult to make Maxam-Gilbert DNA sequencing kit. (Hard to commercialize)
- Read size decrease with incomplete cleavage reactions.
Why Maxam-Gilbert sequencing is now extinct ?
- Even though Sanger sequencing is still widespread, Maxam-Gilbert sequencing has been forgotten. So, you may be surprised to know that when both methods were discovered, Maxam-Gilbert was the most popular. This was because scientists could use purified DNA directly, while the initial Sanger method required cloning for the start of each read
- This method, although based on very simple principles, came with a whole lot of trouble. First, it was time consuming. And that was supposing that everything went well on the first try. A lot of steps in the method could cause problems: the radioactive labeling process, the cleavage reactions, the gel set up, the electrophoresis, and the X-ray film developer. Using this method you could only confirm about 200–300 bases of DNA every few days!
- Maxam-Gilbert sequencing also required working with large amounts of radioactive material and working closely with hydrazine, which is a known neurotoxin. The development of other techniques, and the simplification of Sanger sequencing, caused chemical sequencing to lose its appeal. With the birth of next-generation sequencing, Maxam-Gilbert sequencing is almost extinct and many are claiming the same will happen to Sanger sequencing.
二、Sanger Sequencing
Principles
Sanger sequencing: Chain-terminating method of DNA sequencing
Dideoxy nucleotide mediated chain-terminating method
- 2’,3’-dideoxynucleotide triphosphate (ddNTPs)
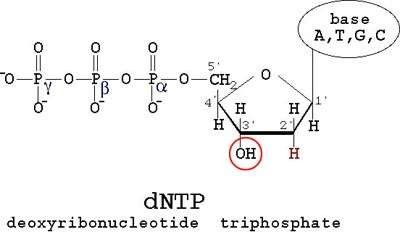
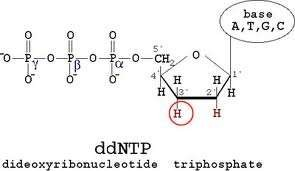
They terminate DNA chain elongation as…
- Cannot form a phosphodiester bond with the next deoxynucleotide
- ddNTPs are the terminator molecules…
Each ddNTP has label for different color fluorescence
1. Ingredients for Sanger Sequencing
- The template DNA to be sequenced
- A DNA polymerase enzyme – A thermo stable form of DNA polymerase, enable doing cycles of reaction in PCR machine.
- The primer, which is a short piece of single-stranded DNA that binds to the template DNA and acts as a “starter” for the polymerase
- The four DNA nucleotides (dATP, dTTP, dCTP, dGTP)
- Dideoxy, or chain-terminating, versions of all four nucleotides (ddATP, ddTTP, ddCTP, ddGTP), each labeled with a different color of dye
- Proportion of the dNTPs and ddNTPs is 100 : 1, Why?
- Means that 1% of the ddNTPs are fluorescence labeled terminator molecules
- If more ddNTPs are included into the reaction, the majority of the growing DNA chain would terminate quickly, reduce the ability to get signal from long enough reads.
- About 500 bp is maximal
2. The Machine Used in Sanger Sequencing
ABI377:

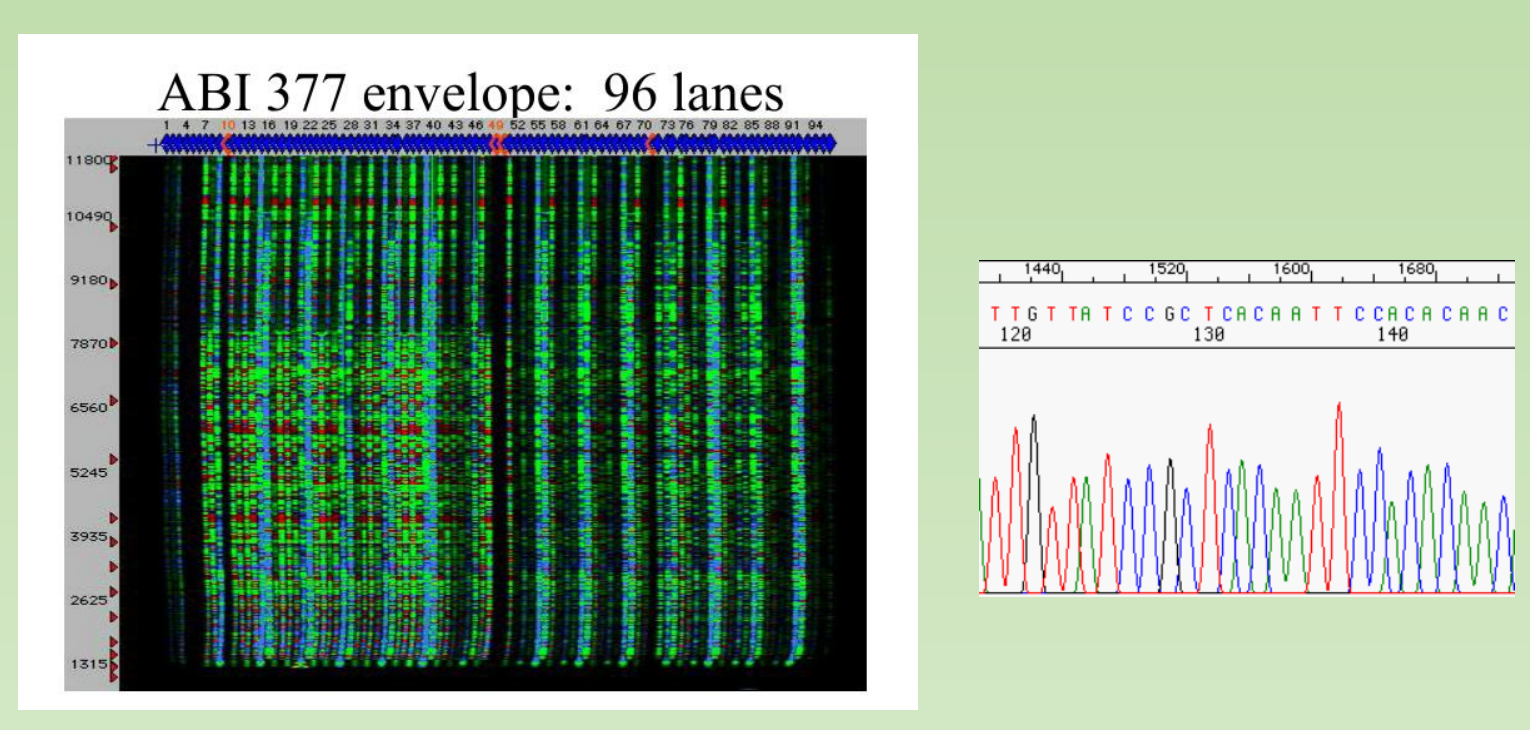
ABI3730, high-throughput Sanger sequencing :

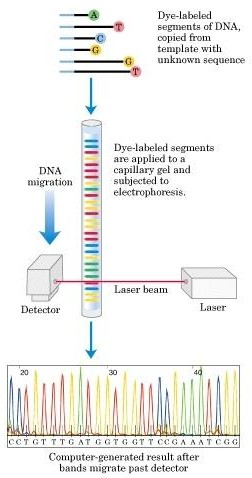
Many Systems for Sanger Automation:

一、Next Generation Sequencing
Illumina
Typical Illumina Setup:

Illumina Outline: The key steps
- Making a sequencing Library
- Cluster generation
- Sequence by synthesis (SBS)
- Data analysis
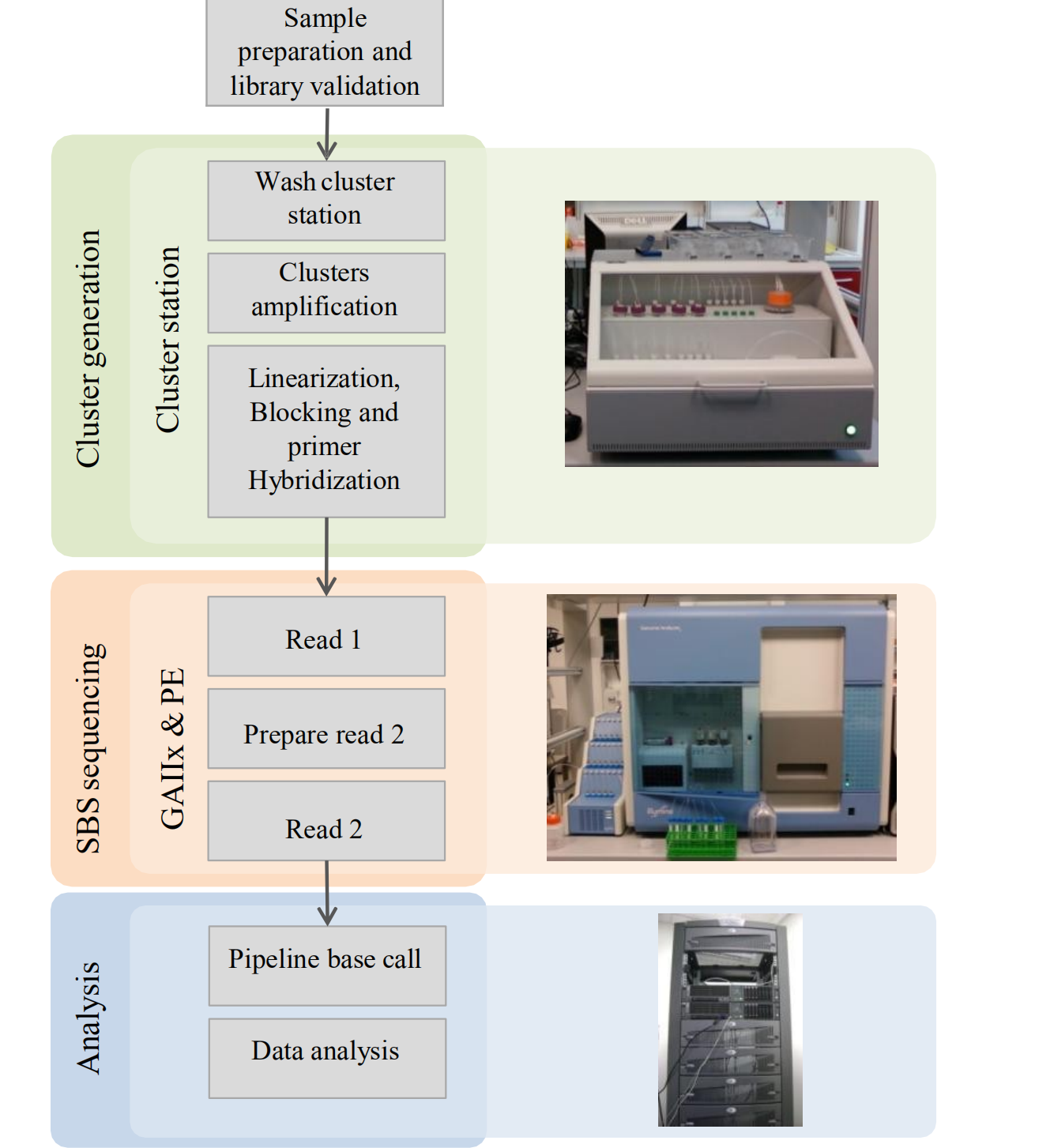
Key Steps
1. Making a Sequencing library


Making a sequencing Library: The key steps
- Add sequencing adaptors to the both end of insert DNA (target DNA).
- The sequencing adaptor enables PCR amplification of the library.
- The sequencing adaptor enables anchoring of the library to the flow cell and the following cluster generation
2. Cluster generation
- Apply the library to the flowcell.
- Cluster generation by solid phase PCR (bridge amplification)
- Each single molecule of DNA in the original sequencing library form a “cluster” on the surface of the flowcell through amplification. (1 million copy of original copy)
3. Sequence by synthesis (SBS)
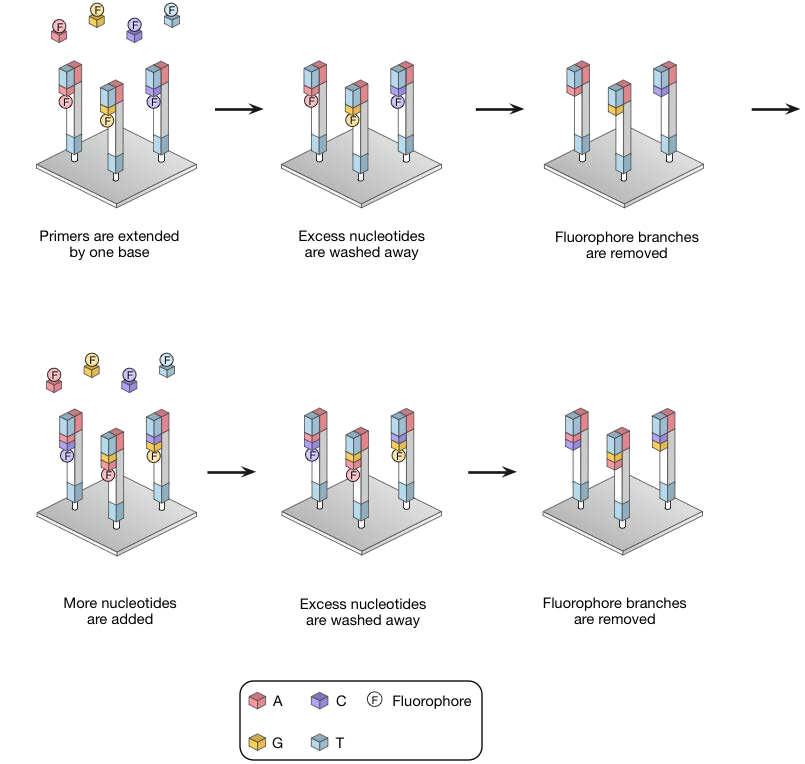
- Fluorescent labeled Reversible terminators are incorporated into the growing DNA chain
- Only a single fluorescent color is used, so each of the four bases must be added in a separate cycle of DNA synthesis and imaging
- Imaging to record the signal
- Next nucleotide is then being added.
4. Data analysis
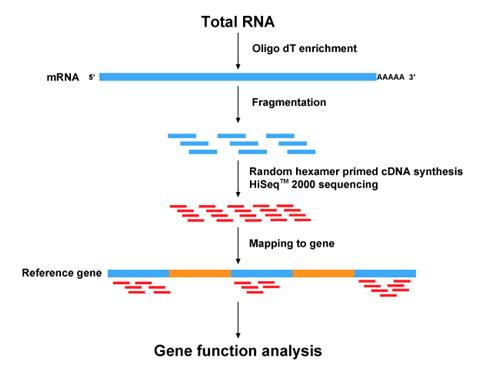
- Initial Data QC
- Processing of raw data (remove low quality reads and trim adaptor, remove PCR duplicate, etc)
- Mapping to the reference genome
- Downstream analysis
Comparison to Sanger Sequencing
| Sanger Sequencing | NGS (Next Generation Sequencing) | |
|---|---|---|
| Benefits | 1. Fast, cost-effective sequencing for low numbers of targets (1–20 targets) 2. Custom friendly 3. Accurate 4. Long read length (500bp) |
1. Higher sequencing depth enables higher sensitivity (down to 1%) 2. Higher mutation resolution 3. More data produced with the same amount of input DNA 4. Higher sample throughput |
| Challenges | 1. Low sensitivity (limit of detection ~15–20%) 2. Not as cost-effective for high numbers of targets (> 20 targets) 3. Low scalability due to increasing sample input requirements |
1. Less cost-effective for sequencing low numbers of targets (1–20 targets) 2. Time-consuming for sequencing low numbers of targets (1–20 targets) |
四、Third Generation Sequencing - Long Read Sequencing
Third generation sequencing- long read sequencing
Third-generation sequencing (also known as long-read sequencing) is a class of DNA sequencing methods currently under active developments
Since eukaryotic genomes contain many repetitive regions, a major limitation to sanger or illumina sequencing methods is the length of reads it produces.

PACBIO: Pacific Bioscience sequencing
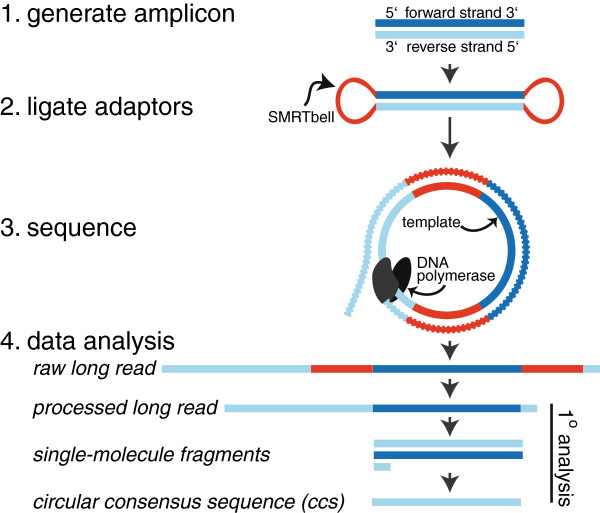
- SMRT(Single-Molecule Real Time Sequencing)
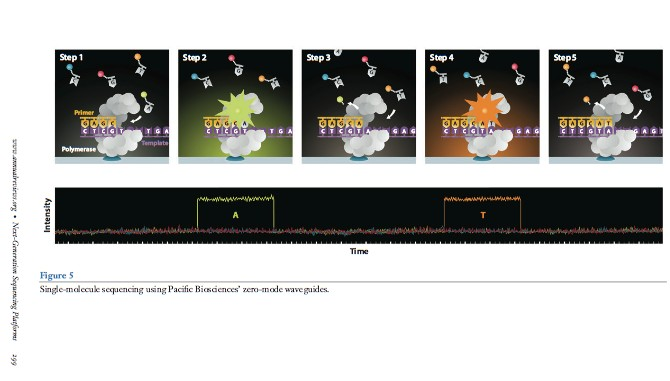
Zero-mode waveguides:
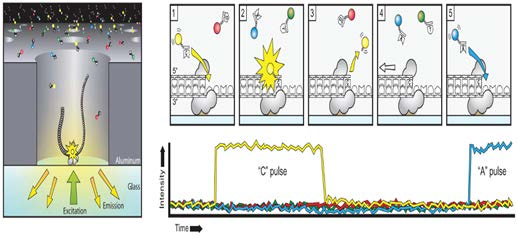
SMRT flow cell, there are thousands of tiny pores called zero-mode waveguides (ZMWs)
Each ZMW represents one reaction chamber and at the bottom of the chamber sits (immobilized) the DNA polymerase machinery.
The pore size (~70nm in diameter, 100nm in depth) is too small for light to pass through easily, thus creating an observation volume that is small enough to observe only a single nucleotide being incorporated and each base is added
Sequence by synthesize. The fluorescent tags are cleaved-off after the incorporation of the nucleotide
work like a giant microscope that can literally “see” DNA synthesis in real time! 1000bp/sec
Can read 20kb long reads. But with lower input and more expensive
- 80Gbp (20kb/read) = 4x10^6 reads = 4 million reads for the whole flow cell.
- 120Gbp (200bp/read) = 120X10^9/ 2x10^2 = 600 Million reads for the whole flow cell
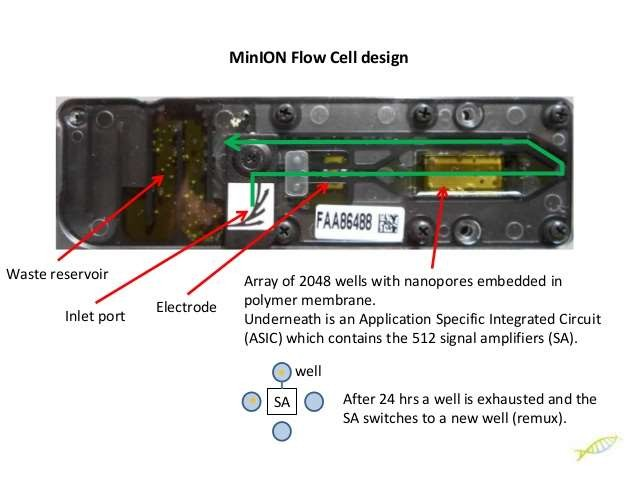
Nanopore Sequencing

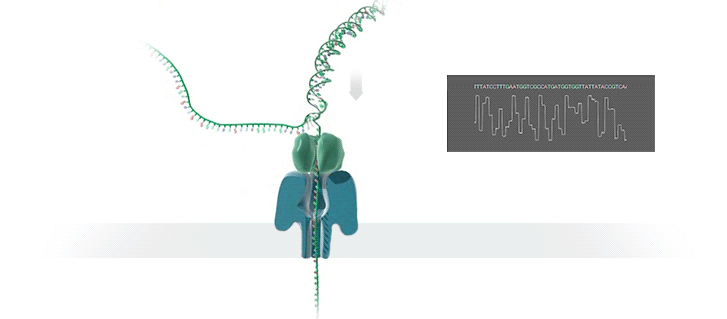
- A protein nanopore is set in an electrically resistant polymer membrane. An ionic current is passed through the nanopore by setting a voltage across this membrane. If an analyte passes through the pore or near its aperture, this event creates a characteristic disruption in current (as shown in the diagram below). Measurement of that current makes it possible to identify the molecule in question
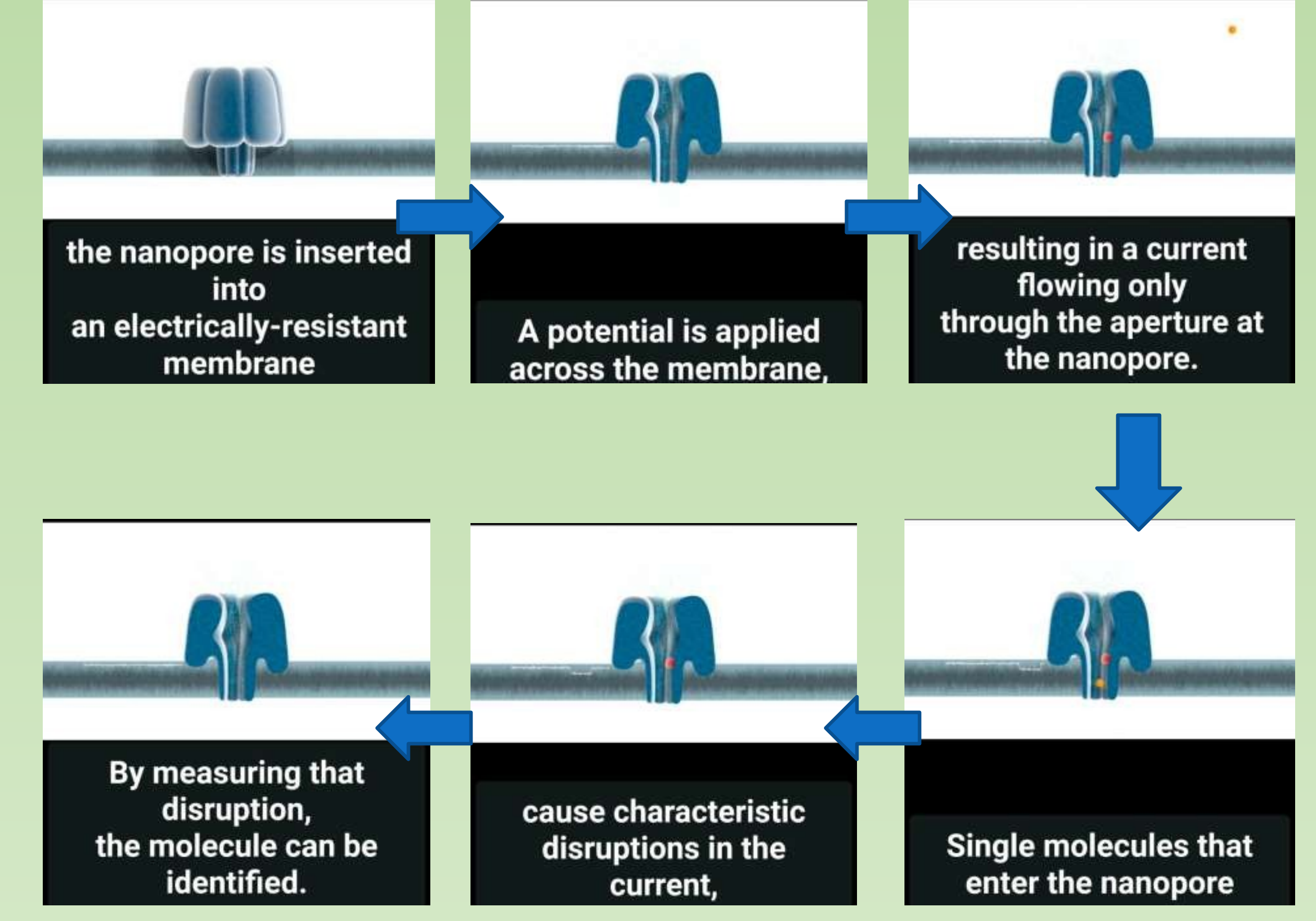
Nanopore
- A protein nanopore is set in an electrically-resistant polymer membrane
Array of microscaffolds
- Each microscaffold supports a membrane and embedded nanopore. The array keeps the multiple nanopores stable during shipping and usage
Sensor chip
- Each microscaffold corresponds to its own electrode that is connected to a channel in the sensor array chip. Sensor arrays may be manufactured with any number of channels
The idea of sample multiplexing:
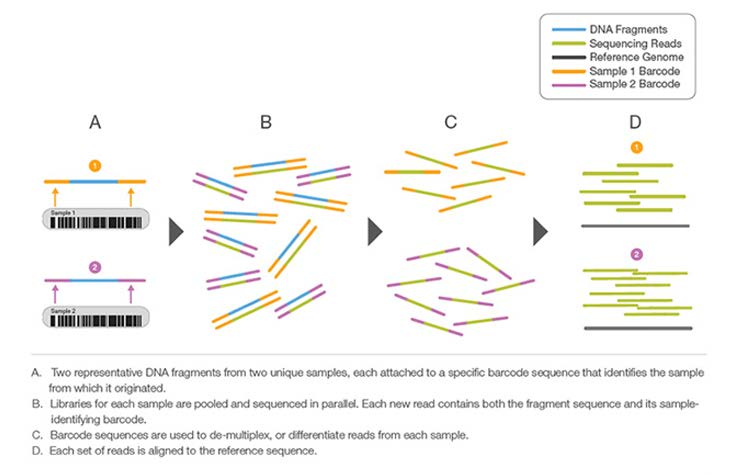
Multiplex Sequencing Highlights
- Fast High-Throughput Strategy: Large sample numbers can be simultaneously sequenced during a single experiment
- Cost-Effective Method: Sample pooling improves productivity by reducing time and reagent use
- Simplified Analysis: Automatic sample identification with “barcodes” using Illumina data analysis softwares
Comparing different sequencing strategies
| Sanger Sequencing First generation | Illumina sequencing Second generation | PACBIO sequencing Third generation | Nanopore sequencing Fourth generation |
|---|---|---|---|
| 1. Dideoxy chain termination 2. Accurate 3. No signal amplification 4. Long read length 500bp 5. Cheap and practical for normal gene cloning |
1. Sequencing by synthesis (Cluster generation/reversible Terminator/imaging are innovations) 2. Involve amplification, accurate at bulk level (not accurate on individual reads) 3. Highest through-put so far 4. Relatively short read length (100-300bp) 5. Ideal for genomic studies |
1. Sequencing by synthesis 2. Anchor the DNA polymerase on chip 3. Long read length (20Kb) due to no amplification, improved imaging technique and rolling cycle sequencing of same molecule 4. Not as high through-put but can provide valuable information 5. Often combined with illumina-seq |
1. Direct sequencing DNA or RNA molecule 2. In theory no limit on DNA length 3. Smallest instrument 4. Lowest instrument cost 5. Highest error rates 6. Often combined with illumina-seq |